Bioinspired Polymer-Bound Organocatalysts for Direct Asymmetric Aldol Reaction: Experimental and Computational Studies



Abstract
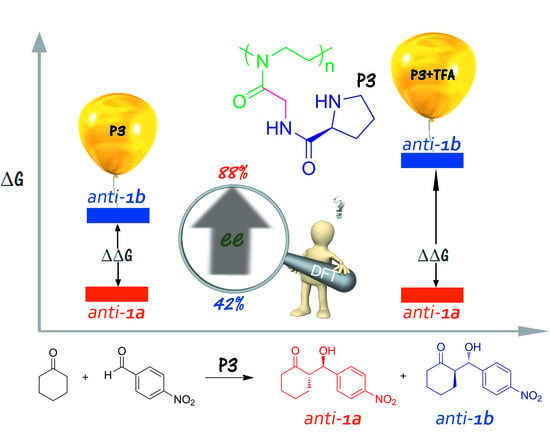
:
1. Introduction
2. Results and Discussion
2.1. Catalyst Synthesis and Structural Characterization
2.2. Evaluation of Catalytic Activity
2.3. Mechanistic Studies Based on DFT Calculations
2.4. Substrate Scope of Aldol Reactions and Recycling Experiments
3. Materials and Methods
3.1. Materials
3.2. Characterization
3.3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- List, B.; Lerner, R.A.; Barbas, C.F., III. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Cozzi, F. Immobilization of organic catalysts: When, why, and how. Adv. Synth. Catal. 2006, 348, 1367–1390. [Google Scholar] [CrossRef]
- Benaglia, M.; Puglisi, A.; Cozzi, F. Polymer-supported organic catalysts. Chem. Rev. 2003, 103, 3401–3430. [Google Scholar] [CrossRef]
- Yin, Y.; Dong, Z.; Luo, Q.; Liu, J. Biomimetic catalysts designed on macromolecular scaffolds. Prog. Polym. Sci. 2012, 37, 1476–1509. [Google Scholar] [CrossRef]
- Breslow, R.; Bandyopadhyay, S.; Levine, M.; Zhou, W. Water exclusion and enantioselectivity in catalysis. Chembiochem 2006, 7, 1491–1496. [Google Scholar] [CrossRef]
- Font, D.; Sayalero, S.; Bastero, A.; Jimeno, C.; Pericàs, M.A. Toward an artificial aldolase. Org. Lett. 2008, 10, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Benaglia, M.; Celentano, G.; Cozzi, F. Enantioselective aldol condensations catalyzed by poly(ethylene glycol)-supported proline. Adv. Synth. Catal. 2001, 343, 171–173. [Google Scholar] [CrossRef]
- Akagawa, K.; Sakamoto, S.; Kudo, K. Direct asymmetric aldol reaction in aqueous media using polymer-supported peptide. Tetrahedron Lett. 2005, 46, 8185–8187. [Google Scholar] [CrossRef]
- Tang, Z.; Iida, H.; Hu, H.Y.; Yashima, E. Remarkable enhancement of the enantioselectivity of an organocatalyzed asymmetric henry reaction assisted by helical poly(phenylacetylene)s bearing cinchona alkaloid pendants via an amide linkage. ACS Macro Lett. 2012, 1, 261–265. [Google Scholar] [CrossRef]
- Karjalainen, E.; Izquierdo, D.F.; Martí-Centelles, V.; Luis, S.V.; Tenhu, H.; García-Verdugo, E. An enzymatic biomimetic system: Enhancement of catalytic efficiency with new polymeric chiral ionic liquids synthesised by controlled radical polymerization. Polym. Chem. 2014, 5, 1437–1446. [Google Scholar] [CrossRef]
- Julia, S.; Guixer, J.; Masana, J.; Rocas, J.; Colonna, S.; Annuziata, R.; Molinari, H. Synthetic enzymes part 2: Catalytic asymmetric epoxidation by means of polyamino-acids in a triphase system. J. Chem. Soc. Perkin Trans. 1 1982, 1317–1324. [Google Scholar] [CrossRef]
- Itsuno, S.; Sakakura, M.; Ito, K. Polymer-supported poly(amino acids) as new asymmetric epoxidation catalyst of α,β-unsaturated ketones. J. Org. Chem. 1990, 55, 6047–6049. [Google Scholar] [CrossRef]
- Davie, E.A.C.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric catalysis mediated by synthetic peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.R.; Roberts, S.M. Oligopeptides as catalysts for asymmetric epoxidation. Pept. Sci. 2006, 84, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Aoi, K.; Okada, M. Polymerization of oxazolines. Prog. Polym. Sci. 1996, 21, 151–208. [Google Scholar] [CrossRef]
- Hoogenboom, R. Poly(2-oxazoline)s: A polymer class with numerous potential applications. Angew. Chem. Int. Ed. 2009, 48, 7978–7994. [Google Scholar] [CrossRef] [PubMed]
- Schlaad, H.; Diehl, C.; Gress, A.; Meyer, M.; Demirel, A.L.; Nur, Y.; Bertin, A. Poly(2-oxazoline)s as smart bioinspired polymers. Macromol. Rapid Commun. 2010, 31, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, R.; Schlaad, H. Bioinspired poly(2-oxazoline)s. Polymers 2011, 3, 467–488. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, H.; Zhou, L.; Hu, F.; Xie, S.; Jiang, L. Novel poly(2-oxazoline)s with pendant l-prolinamide moieties as efficient organocatalysts for direct asymmetric aldol reaction. Catal. Sci. Technol. 2016, 6, 6739–6749. [Google Scholar] [CrossRef]
- Chen, F.; Huang, S.; Zhang, H.; Liu, F.; Peng, Y. Proline-based dipeptides with two amide units as organocatalyst for the asymmetric aldol reaction of cyclohexanone with aldehydes. Tetrahedron 2008, 64, 9585–9591. [Google Scholar] [CrossRef]
- Miura, T.; Imai, K.; Ina, M.; Tada, N.; Imai, N.; Itoh, A. Direct asymmetric aldol reaction with recyclable fluorous organocatalyst. Org. Lett. 2010, 12, 1620–1623. [Google Scholar] [CrossRef]
- Fuentes de Arriba, A.L.; Simon, L.; Raposo, C.; Alcazar, V.; Sanz, F.; Muniz, F.M.; Moran, J.R. Imidazolidinone intermediates in prolinamide-catalyzed aldol reactions. Org. Biomol. Chem. 2010, 8, 2979–2985. [Google Scholar] [CrossRef]
- Kobayashi, S. Ethylenimine polymers. Prog. Polym. Sci. 1990, 15, 751–823. [Google Scholar] [CrossRef]
- Wenker, H.J. Syntheses from ethanolamine V. synthesis of Δ2-oxazoline and of 2,2′-Δ2-dioxazoline. J. Am. Chem. Soc. 1938, 60, 2152–2153. [Google Scholar] [CrossRef]
- Hu, F.; Xie, S.; Jiang, L.; Shen, Z. Living cationic ring-opening polymerization of 2-oxazolines initiated by rare-earth metal triflates. RSC Adv. 2014, 4, 59917–59926. [Google Scholar] [CrossRef]
- Torii, H.; Nakadai, M.; Ishihara, K.; Susumu, S.; Yamamoto, H. Asymmetric direct aldol reaction assisted by water and a proline-derived tetrazole catalyst. Angew. Chem. Int. Ed. 2004, 43, 1983–1986. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, L. Mechanisms and reactivity differences of proline-mediated catalysis in water and organic solvents. Catal. Sci. Technol. 2016, 6, 3378–3385. [Google Scholar] [CrossRef]
- Doyagüez, E.G.; Parra, F.; Corrales, G.; Fernández-Mayoralas, A.; Gallardo, A. New hydroxyproline based methacrylic polybetaines: Synthesis, pH sensitivity and catalytic activity. Polymer 2009, 50, 4438–4446. [Google Scholar] [CrossRef]
- Lei, M.; Shi, L.; Li, G.; Chen, S.; Fang, W.; Ge, Z.; Cheng, T.; Li, R. Dipeptide-catalyzed direct asymmetric aldol reactions in the presence of water. Tetrahedron 2007, 63, 7892–7898. [Google Scholar] [CrossRef]
- Haraguchi, N.; Kiyono, H.; Takemura, Y.; Itsuno, S. Design of main-chain polymers of chiral imidazolidinone for asymmetric organocatalysis application. Chem. Commun. 2012, 48, 4011–4013. [Google Scholar] [CrossRef]
- Tuchman-Shukron, L.; Miller, S.J.; Portnoy, M. Polymer-supported enantioselective bifunctional catalysts for nitro-michael addition of ketones and aldehydes. Chem. Eur. J. 2012, 18, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Tan, R.; Zhao, L.; Yin, D. l-Proline supported on ionic liquid-modified magnetic nanoparticles as a highly efficient and reusable organocatalyst for direct asymmetric aldol reaction in water. Green Chem. 2013, 15, 2422–2433. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Salvo, A.M.P.; Giacalone, F.; Agrigento, P.; Noto, R. Enhanced activity and stereoselectivity of polystyrene-supported proline-based organic catalysts for direct asymmetric aldol reaction in water. Eur. J. Org. Chem. 2009, 5437–5444. [Google Scholar] [CrossRef]
- Bertelsen, S.; Jorgensen, K.A. Organocatalysis—After the gold rush. Chem. Soc. Rev. 2009, 38, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Bassan, A.; Zou, W.; Reyes, E.; Himo, F.; Cordova, A. The origin of stereoselectivity in primary amino acid catalyzed intermolecular aldol reactions. Angew. Chem. Int. Ed. 2005, 44, 7028–7032. [Google Scholar] [CrossRef] [PubMed]
- Bahmanyar, S.; Houk, K.N. Transition states of amine-catalyzed aldol reactions involving enamine intermediates: Theoretical studies of mechanism, reactivity, and stereoselectivity. J. Am. Chem. Soc. 2001, 123, 11273–11283. [Google Scholar] [CrossRef] [PubMed]
- Bahmanyar, S.; Houk, K.N.; Martin, H.J.; List, B. Quantum mechanical predictions of the stereoselectivities of proline-catalyzed asymmetric intermolecular aldol reactions. J. Am. Chem. Soc. 2003, 125, 2475–2479. [Google Scholar] [CrossRef]
- Kobayashi, S.; Hachiya, I. Lanthanide triflates as water-tolerant lewis acids activation of commercial formaldehyde solution and use in the aldol reaction of silyl enol ethers with aldehydes in aqueous media. J. Org. Chem. 1994, 59, 3590–3596. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst b | Loading (mol.%) | Time (h) | Yield (%) c | anti/synd | % ee of anti-1 e |
| 1f | (R)-P1 | 10 | 24 | 44 | 80:20 | 91 (34) |
| 2 | (S)-P1 | 10 | 24 | 42 | 80:20 | 87 (16) |
| 3 | P2 | 10 | 48 | ~5 | − | − |
| 4 | P3 | 10 | 12 | 86 | 84:16 | 88 (33) |
| 5 | (R)-P4 | 10 | 12 | 94 | 75:25 | 79 (11) |
| 6 | (S)-P4 | 10 | 12 | 91 | 79:21 | 71 (5) |
| 7 | P5 | 10 | 12 | 9 | 74:26 | 68 (26) |
| 8 | MC1 | 10 | 48 | 29 | 70:30 | 58 (6) |
| 9 | MC2 | 10 | 24 | 22 | 74:26 | 64 (8) |
| 10 | P3 | 5 | 24 | 54 | 82:18 | 89 (27) |
| 11 | P3 | 5 | 48 | 84 | 85:15 | 89 (27) |
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol.%) | TFA (equiv.) | Yield (%) b | anti/sync | % ee of anti-1 d |
| 1 | P3 (10) | 0 | 40 | 72:28 | 42 (5) |
| 2 | P3 (10) | 0.2 | 73 | 77:23 | 73 (48) |
| 3 | P3 (10) | 0.4 | 79 | 79:21 | 77 (53) |
| 4 | P3 (10) | 0.6 | 85 | 81:19 | 80 (38) |
| 5 | P3 (10) | 0.8 | 86 | 84:16 | 88 (33) |
| 6 | P3 (10) | 1 | 33 | 83:17 | 88 (39) |
| 7 e | P3 (5) | 0.8 | 84 | 85:15 | 89 (27) |
| 8 e | P3 (5) | 1.6 | 15 | 79:21 | 82 (17) |
| 9 | P5 (10) | 0 | 77 | 79:21 | 47 (28) |
| 10 | P5 (10) | 0.8 | 9 | 74:26 | 68 (26) |
| Entry | Catalyst | Without TFA | With TFA | Product b | ||
|---|---|---|---|---|---|---|
| TS | ΔΔG (kcal/mol) | TS | ΔΔG (kcal/mol) | |||
| 1 | P3 | TSa1 | 0 | TSc1 | 0 | anti-1a |
| 2 | TSa2 | 2.83 | TSc2 | 3.12 | syn-1a | |
| 3 | TSa3 | 4.27 | TSc3 | 3.60 | syn-1b | |
| 4 | TSa4 | 1.48 | TSc4 | 19.28 | anti-1b | |
| 5 | P5 | TSb1 | 0 | TSd1 | 0 | anti-1a |
| 6 | TSb2 | 3.66 | TSd2 | 2.19 | syn-1a | |
| 7 | TSb3 | 5.39 | TSd3 | 3.14 | syn-1b | |
| 8 | TSb4 | 1.84 | TSd4 | 19.47 | anti-1b | |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | X | R | Product | Yield (%) b | syn/antic | ee (%) d |
| 1 | −CH2CH2− | 2-NO2 | 3a | 86 | 15/85 | 90 |
| 2 | −CH2CH2− | 3-NO2 | 3b | 96 | 18/82 | 91 |
| 3 | −CH2CH2− | 4-CN | 3c | 83 | 30/70 | 88 |
| 4 | −CH2CH2− | 4-CO2CH3 | 3d | 61 | 13/87 | 80 |
| 5 | −CH2CH2− | H | 3e | <5 | − | − |
| 6 | −CH2CH2− | 4-OCH3 | 3f | <5 | − | − |
| 7 | −CH2O− | 4-NO2 | 3g | 97 | 15/85 | 94 |
| 8 | −CH2− | 4-NO2 | 3h | 70 | 25/75 | 72 |
| Entry | Cycle | Yield (%) b | syn/antic | ee (%) d |
|---|---|---|---|---|
| 1 | 1 | 86 | 24:76 | 88 |
| 2 | 2 | 85 | 20:80 | 90 |
| 3 | 3 | 85 | 19:81 | 91 |
| 4 | 4 | 82 | 16:84 | 89 |
| 5 | 5 | 79 | 13:87 | 92 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, G.; Ling, J.; Hu, F.; Liu, K.; Ye, L.; Jiang, L. Bioinspired Polymer-Bound Organocatalysts for Direct Asymmetric Aldol Reaction: Experimental and Computational Studies. Catalysts 2019, 9, 398. https://doi.org/10.3390/catal9050398
Du G, Ling J, Hu F, Liu K, Ye L, Jiang L. Bioinspired Polymer-Bound Organocatalysts for Direct Asymmetric Aldol Reaction: Experimental and Computational Studies. Catalysts. 2019; 9(5):398. https://doi.org/10.3390/catal9050398
Chicago/Turabian StyleDu, Ganhong, Jun Ling, Fangyu Hu, Keyuan Liu, Long Ye, and Liming Jiang. 2019. "Bioinspired Polymer-Bound Organocatalysts for Direct Asymmetric Aldol Reaction: Experimental and Computational Studies" Catalysts 9, no. 5: 398. https://doi.org/10.3390/catal9050398
APA StyleDu, G., Ling, J., Hu, F., Liu, K., Ye, L., & Jiang, L. (2019). Bioinspired Polymer-Bound Organocatalysts for Direct Asymmetric Aldol Reaction: Experimental and Computational Studies. Catalysts, 9(5), 398. https://doi.org/10.3390/catal9050398