1. Introduction
Supramolecular catalysis in confined space is today a consolidated branched of the supramolecular chemistry, which is inspired by nature, in particular by natural enzymes. The goal of this approach is to create a micro- or nano-environment having features different from the bulk solution. In this confined space, reactivity is different with respect to the normal conditions in solution, and thus the rate of the reaction, as well as the products, can be different with respect to classic catalysis.
Enzymes provide a pocket (the active site) able to recognize the specific substrate via multiple interactions (such as hydrophobic interactions, hydrogen bonds, Lewis acid–base interactions), increasing the reaction rate, leading to stereo- and regioselective reactions. The modern supramolecular catalysts take inspiration from nature, designing artificial nanozymes able to include and transform specific substrates into the desired products.
A crucial point of the supramolecular catalysis in confined space is the proximity effect: reagents, hosted in a supramolecular capsule, are forced to be close to each other, removing the solvent molecules and thus leading to an increase of the activity/reactivity. In addition, the inclusion into a restricted space leads the reagent molecules to react each other with specific geometric constraints, leading to reaction products with regio- and stereoselectivity different from the normal conditions in solution.
A fundamental step in order to create an efficient supramolecular nanocatalyst is the design of the supramolecular host. Today, supramolecular chemistry offers many synthetic macrocycles which can be used to create a confined nanospace: resorcinarenes [
1,
2], cavitands [
2,
3,
4,
5,
6], metallocages [
7], and capsules assembled by hydrophobic interactions [
8]. Although different supramolecular assemblies based on the abovementioned hosts have been studied for molecular recognition, regarding catalysis, only supramolecular containers assembled via hydrogen bonding and metal coordination have been reported to date. In fact, the use of oligomeric hosts such as cucurbiturils, cyclodextrins, crown ethers, calixarenes, and carcerands is complicated due to their relatively small cavity dimensions and difficulty in obtaining larger receptors.
In 2015, a comprehensive review reported the applications of some nanocontainers in the catalytic field [
9]. Thus, this review is focused on the recent catalytic applications to date since 2015.
2. Catalysis into Resorcinarene Hexameric Capsules
Resorcin[4]arenes are synthetic organic compounds able to form a hemispherical structure with a diameter of ca. 10 Å at the upper rim and a hydrophobic cavity with an internal volume of 350 Å
3 that can accommodate many organic molecules [
10]. These macrocycles are synthesized starting from resorcinol or 1,2,3-trihydroxyphenol and an appropriate aldehyde, which addresses the chemical characteristics of the lower rim [
11]. The presence of eight hydroxyl groups on the upper rim opens the possibility of a wide functionalization range and of building supramolecular capsules exploiting hydrogen bonds [
12].
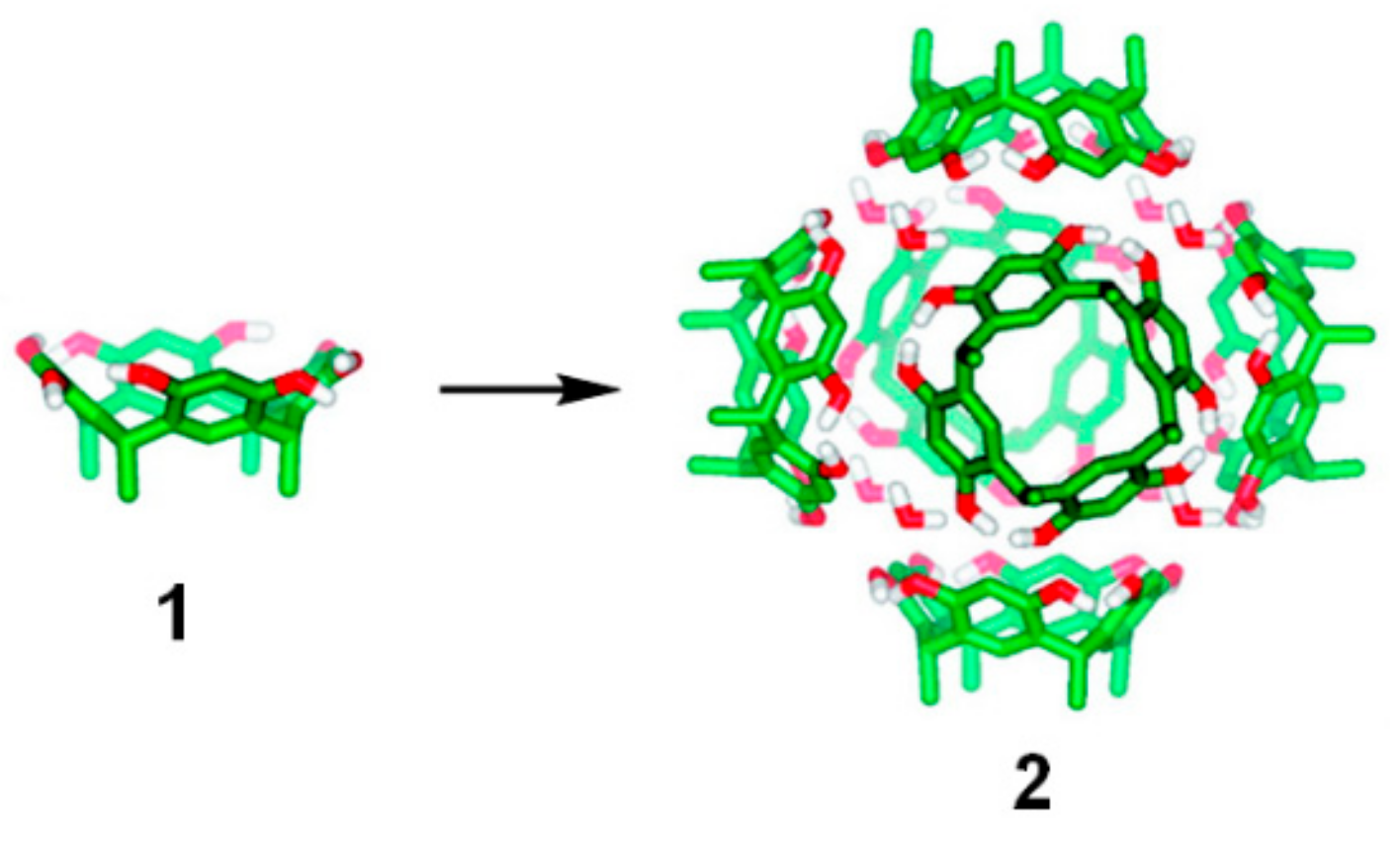
By forming multiple hydrogen bonds, resorcin[4]arene
1 can form hexameric capsules through a self-assembly process first reported by Atwood and coworkers in 1997 (
Figure 1) [
13,
14].
The presence of water molecules is essential to the formation of the supramolecular assembly [
15], which reaches an internal volume of ca. 1400 Å
3, ideal for many organic reactions to be carried out inside. Assembly and molecular recognition properties of hexameric capsule
2 have been extensively investigated in solution [
16,
17,
18]. A comprehensive review collected catalytic applications of resorcinarene capsules until 2014 [
8]; in addition, a recent review by Gaeta and coworkers analyzed some important issues of the catalysis into capsule
2, such as substrate selectivity, transition state stabilization, and acid- and hydrogen-bond-catalyzed reactions [
19]. Furthermore, recently, cyclization reactions inside hexameric capsules have been summarized by Rebek [
20]; thus, here, we have summarized the other recent developments.
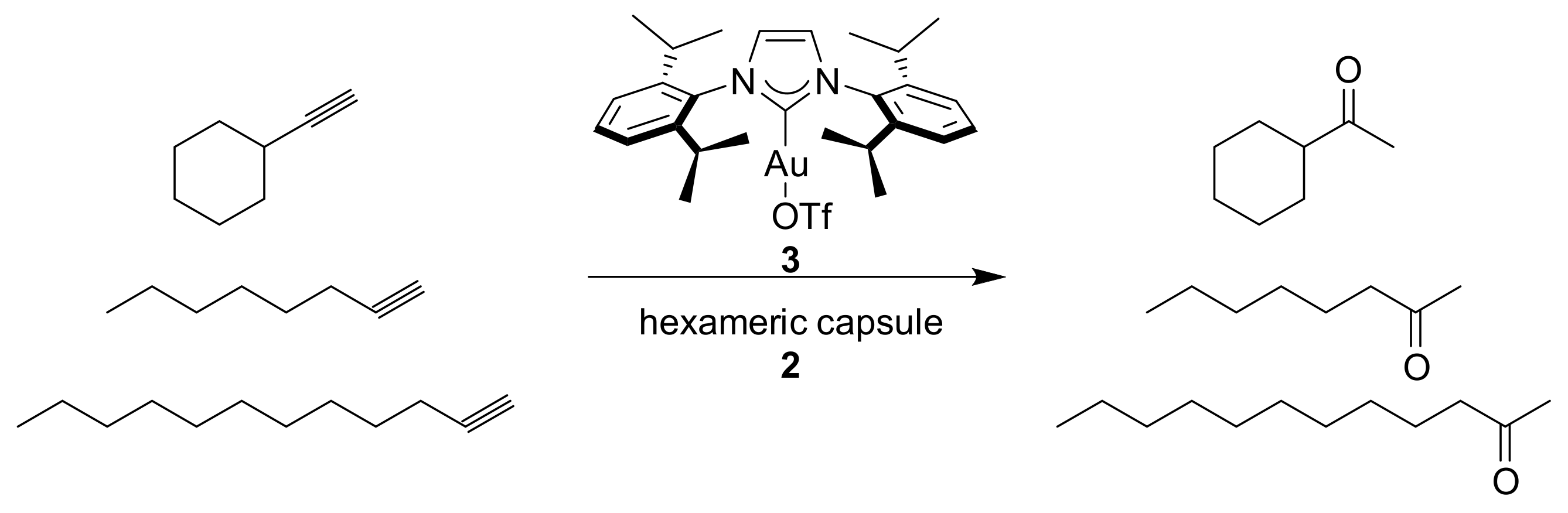
The research group of Prof. Scarso has made a considerable contribution in this field. They reported the use of capsule
2 in the hydration reaction of alkynes [
21]. In particular, they studied the effect of encapsulation of the gold catalyst
3 into
2 on substrate selectivity in the hydration of terminal alkynes (
Scheme 1). The substrates tested differ structurally at a region far from the reactive triple bond.
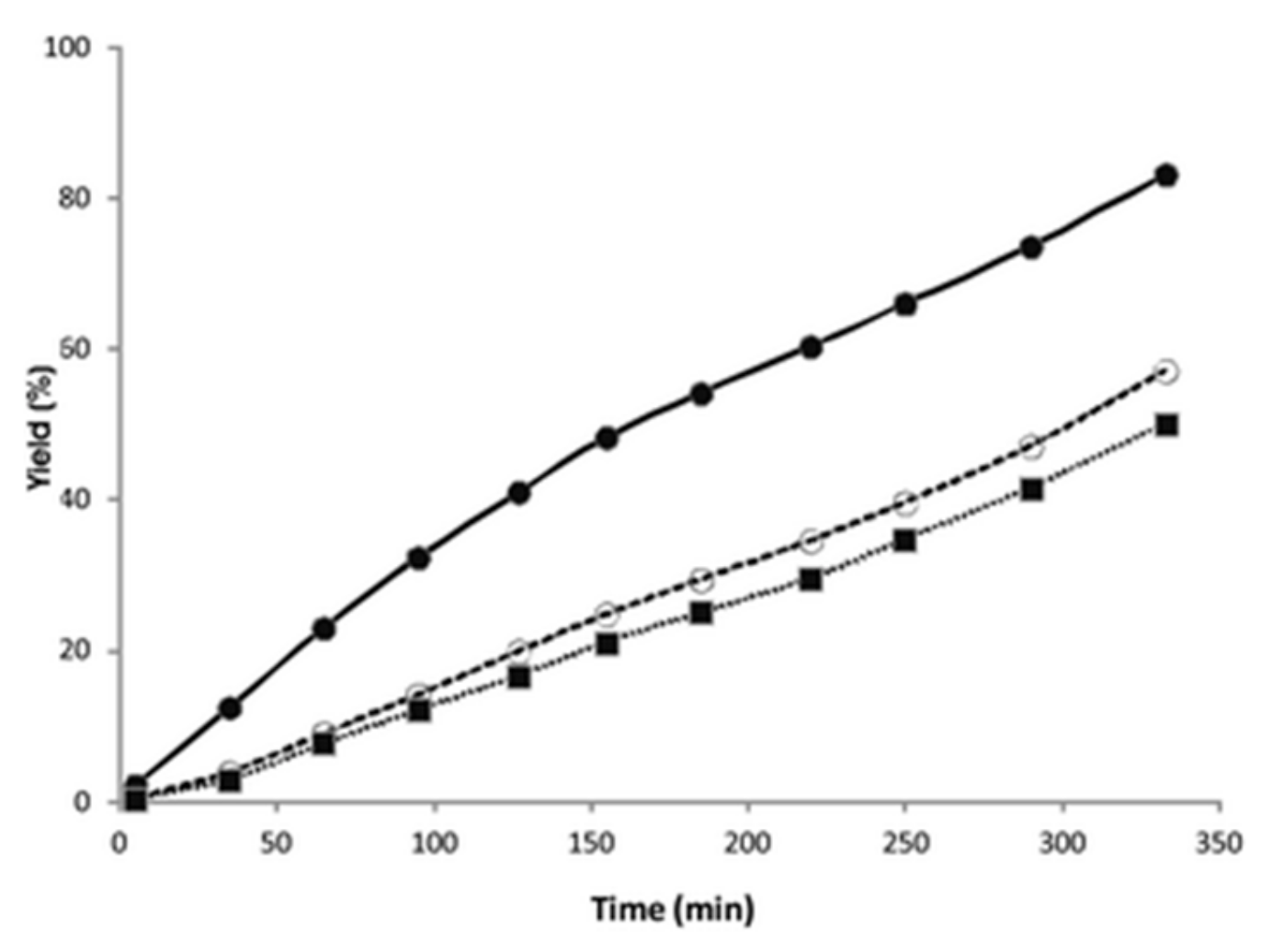
Ethynylcyclohexane, 1-octyne, and 1-dodecyne were tested at 40 °C with 5 mol % of catalyst with and without the capsule. Reactions inside
2 are slower with respect to the free solution, due to the inclusion of the substrates inside the hydrophobic cavity. The reaction profiles showed in
Figure 2 suggest a preference for the cyclic alkyne with respect to the linear one. In particular, the initial rate with ethynylcyclohexane is about three times higher than that obtained with linear substrates, while in the absence of the supramolecular capsule, the reaction rates are similar.
A possible explanation could be the different shape of the cyclic substrate compared to the linear ones. Then, the authors tested cyclic alkynes with different steric dimensions. Inside the capsule, the smaller aromatic alkyne reacted much faster than those of the bigger substrates, while in free solution, the reactivity is the opposite.
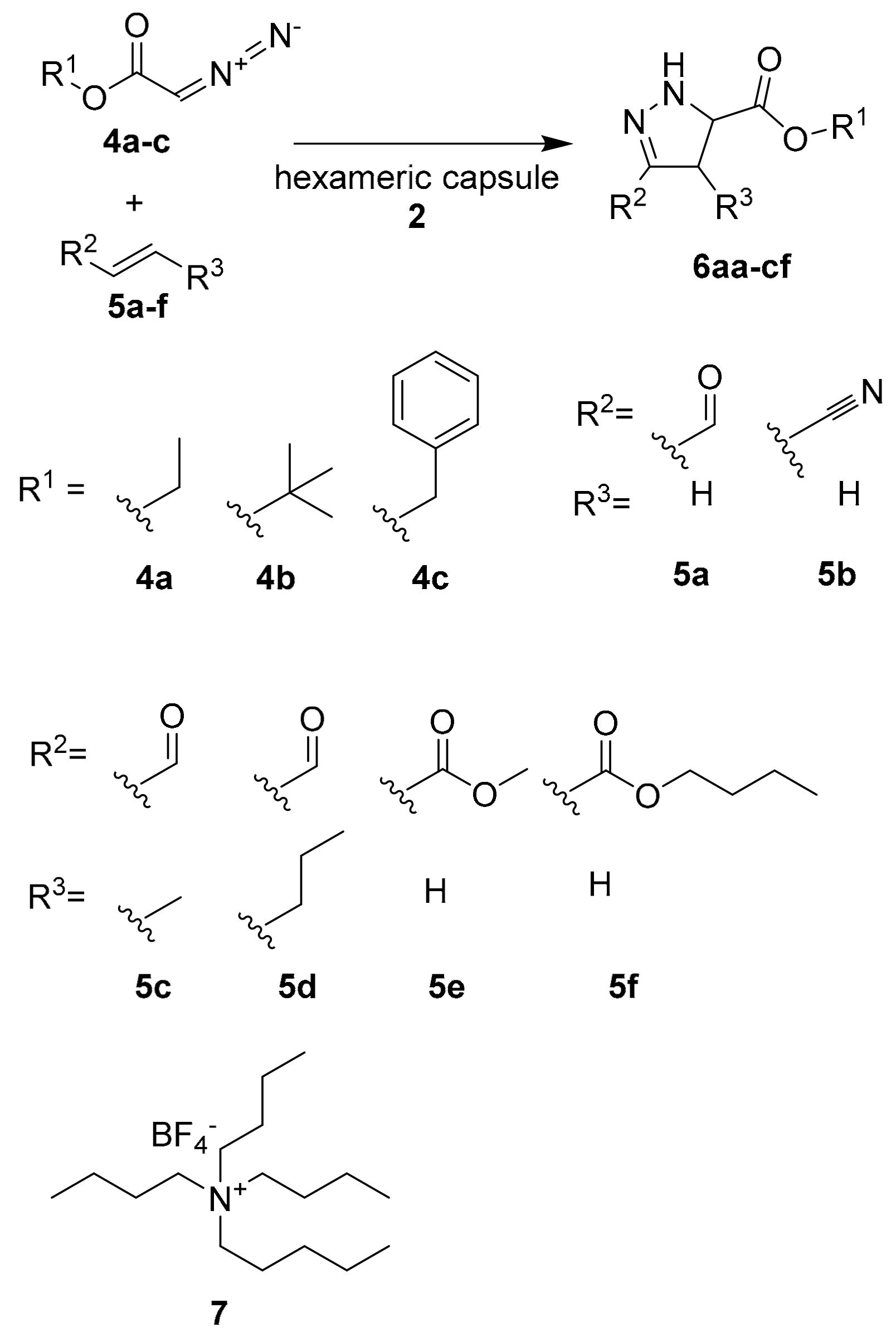
The same research group employed hexameric capsule
2 as the reactor for a dipolar cycloaddition reaction between diazoacetate esters and electron-poor alkenes (
Scheme 2) [
22].
The reaction occurs spontaneously at high concentration levels or can be catalyzed by Lewis acids/bases. When using acrolein as an active electron-poor alkene, the capsule leads to an increase of the yield respect to the free solution reaction (from 12% without capsule to 47% in the presence of the capsule) (see
Table 1, entries 1 and 2). The addition of a large excess of tetrabutylammonium strongly decreases the yield, due to the inclusion of the cation in the cavity (
Table 1, entry 3). In fact, tetrabutylammonium has a higher affinity for the cavity with respect to the reagents. The same trend is observed also with the other substrates, demonstrating that the dipolar cycloaddition reactions occur within the cavity with higher performance compared to in the absence of the supramolecular capsule. Furthermore, the authors extended the screening of substrates to other electron-poor alkenes such as
trans-crotonaldehyde and
trans-2-hexenal. Very interesting are the cases of
5c–
e as substrates: in the absence of a capsule, no reaction occurs, while the presence of capsule
2 leads to a 95% of
5c and 79% yield of
5d–
e. Using bigger substrates, the catalytic activity of the capsule decreases due to the steric hindrance.
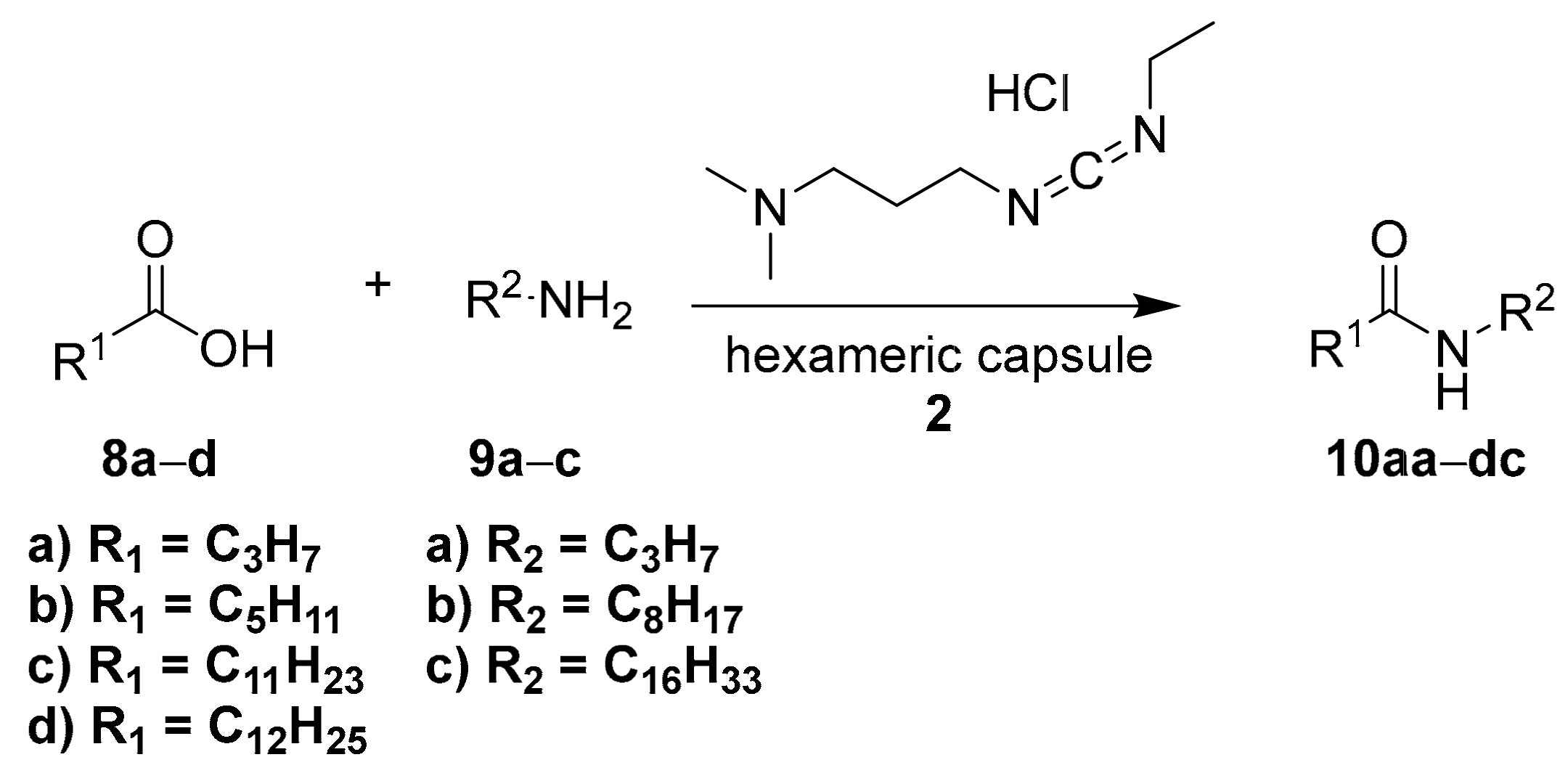
The synthesis of amides driven by a carbodiimide condensing agent is a reaction extremely widespread in organic chemistry. Scarso and coworkers used molecular capsule
2 as the nanoreactor for the synthesis of amides, starting from linear carboxylic acids and primary amines, by using
N-(3-dimethylaminopropyl)-
N′-ethylcarbodiimide hydrochloride (EDAC) as the activating agent (
Scheme 3) [
23].
The hexameric capsule prefers shorter substrates, leading to the formation of the shorter amides, while in the absence of the capsule, the reactivity of all substrates is the same. These data suggest that the driving force for the substrate selectivity is the molecular recognition of the reagents, tuned by the steric effect.
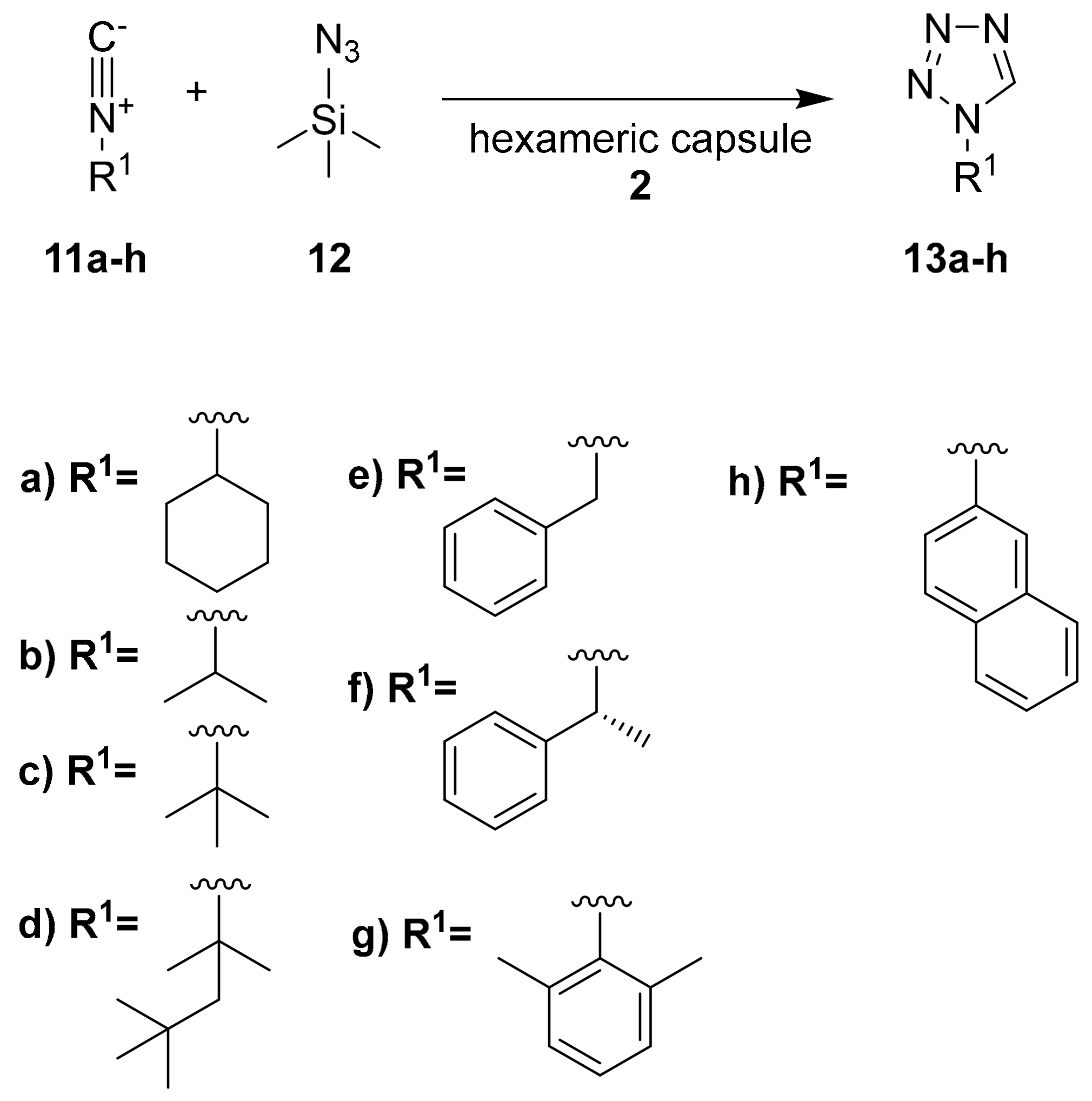
Similar considerations can be related to the synthesis of 1H tetrazoles carried out in capsule
2 (
Scheme 4) [
24].
Table 2 shows the effect of the hexameric capsule in the click reaction using substrate
11a. In particular, no reaction occurs in the absence of the capsule. On the contrary, the presence of 10% of capsule
2 leads to the formation of the desired compound in an almost quantitative yield. The authors also tested the effect of a Bronsted acid (acetic acid) and resorcinol (
Table 2, entries 3 and 4, respectively) in the absence of the hexameric capsule. Bronsted acids, in fact, can catalyze the reaction in normal conditions. However, the obtained yields are lower with respect to the reaction inside the hexameric capsule. As previously reported, the presence of a competitive guest (TEA) inhibits the reaction, leading to a decrease of the yield (
Table 2, entry 5).
The authors tested also other aliphatic and aromatic substrates (
Table 3). In general, the presence of the free capsule leads to higher yields with respect to the occupied cavity (the presence of TEA decreases the yield values). Notably, for aromatic substrates (
11g and
11h), the inhibition of the catalytic activity by using TEA is emphasized.
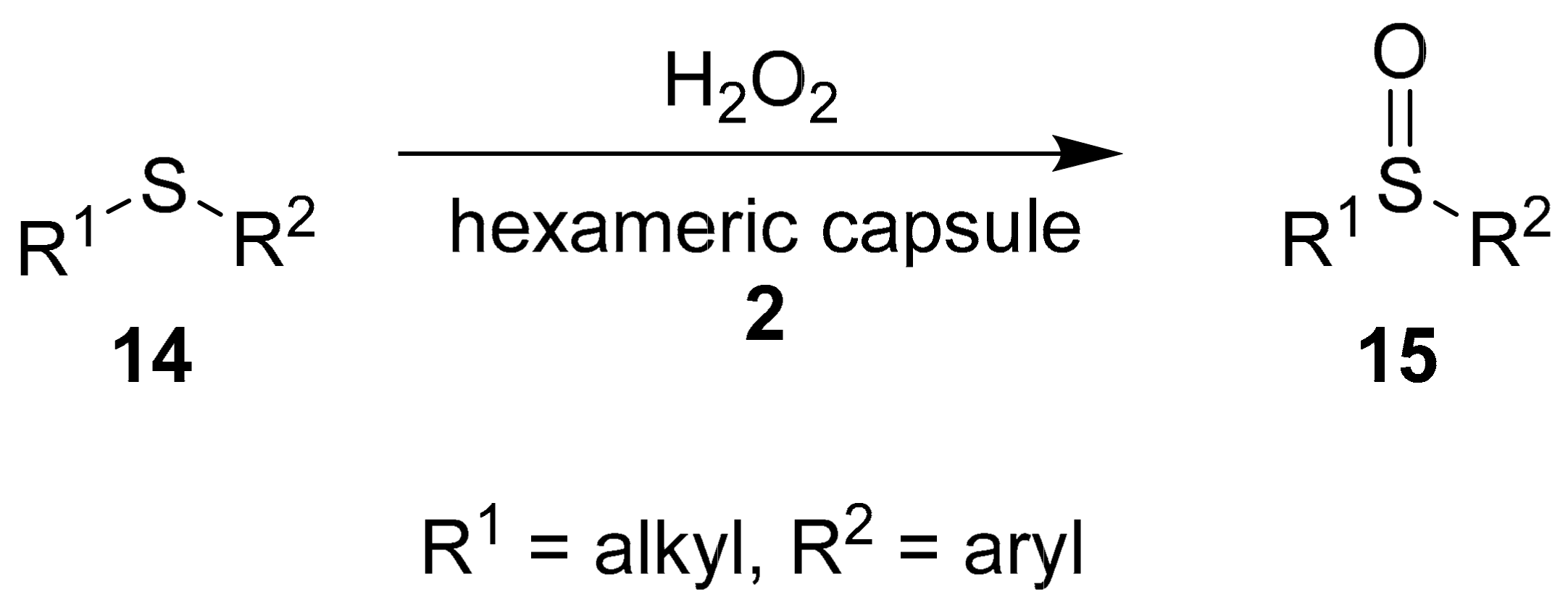
Another important organic reaction catalyzed into hexameric capsule
2 is the sulfoxidation of thioethers, conducted under mild conditions within few hours (
Scheme 5) [
25].
The authors optimized the reaction using dibutyl sulfide as a substrate (
Table 4). In particular, the positive effect of the hexameric capsule is demonstrated by the comparison of entries 1 and 2 in
Table 4. In free solution, a yield of 10% is obtained after 90 min, while within the nano-environment, quantitative amounts of sulfoxide are obtained in 65 min. Due to the possibility of catalyzing the electrophilic oxidation of thioethers with H
2O
2 by the presence of phenols or acids, the authors tested the reaction with acetic acid and resorcinol (
Table 4, entries 3 and 4, respectively). In both cases, although the yields are higher with respect to the uncatalyzed reaction (
Table 4, entry 1), the efficiency of sulfoxidation is lower with respect to the encapsulated oxidation (
Table 4, entry 2). Also, in this case, TEA was used as an inhibitor (
Table 4, entries 5 and 6), leading to a moderate inhibition of the sulfoxidation. The authors assumed that the reaction is favored within the capsule due to the displacement of the water molecules of hexameric capsule
2 by hydrogen peroxide, thus making it more electrophilic (oxidant activation). In addition, the transition state could be stabilized into the capsule by the electron-rich internal surface.
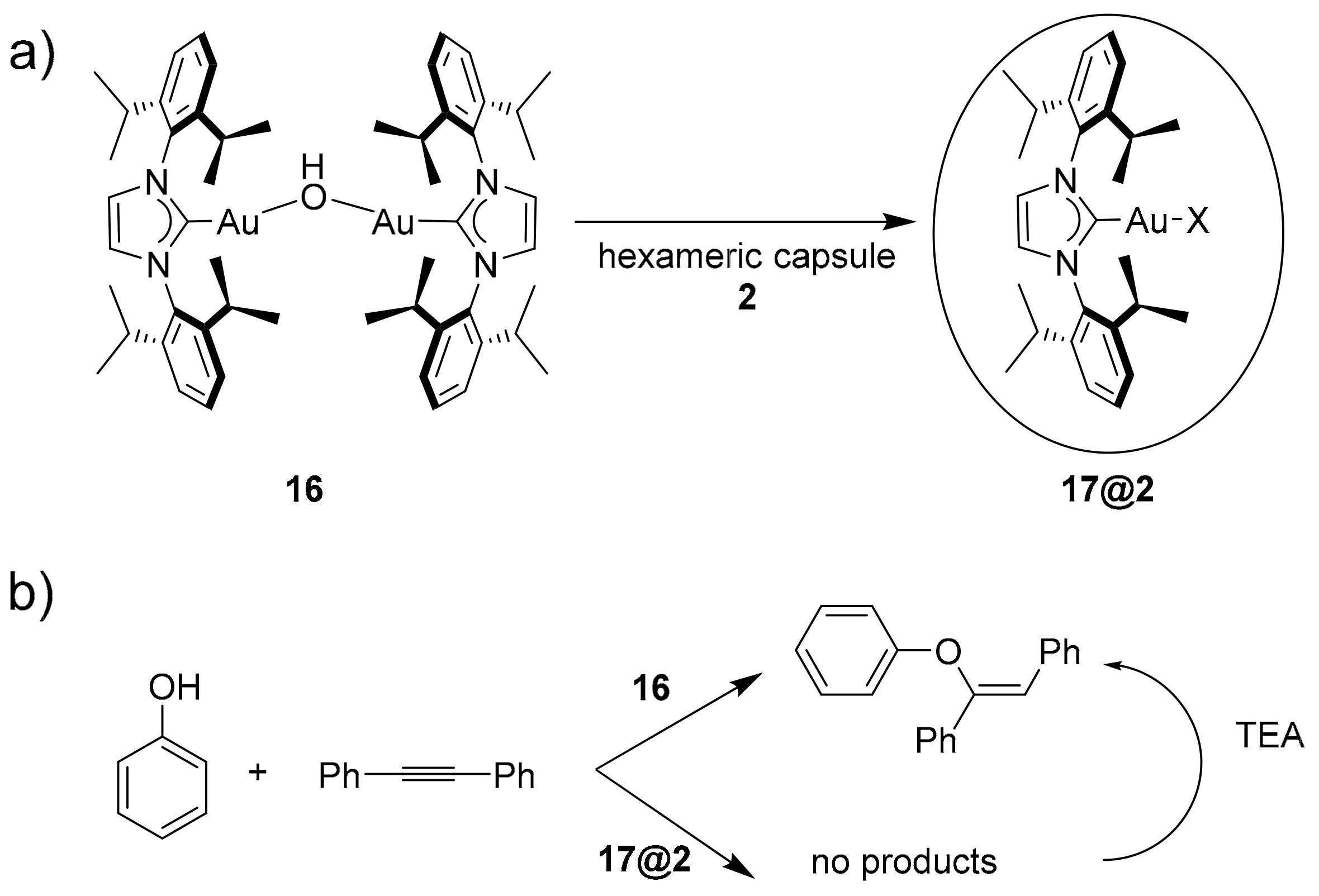
Reek and coworkers exploited the ability of hexameric capsule
2 to inhibit or modify the reactivity of dimeric gold catalyst
16 (
Scheme 6a) [
26]. In particular, the dimeric catalyst
16 leads to the vinyl ether reported in
Scheme 6b after 60 min in an almost quantitative yield. On the contrary, no products were observed after 24 h in the presence of the monomeric gold catalyst within the hexameric capsule
17@2. The addition of TEA as a competitive guest regenerates the original dimeric gold catalyst, leading to the desired compound.
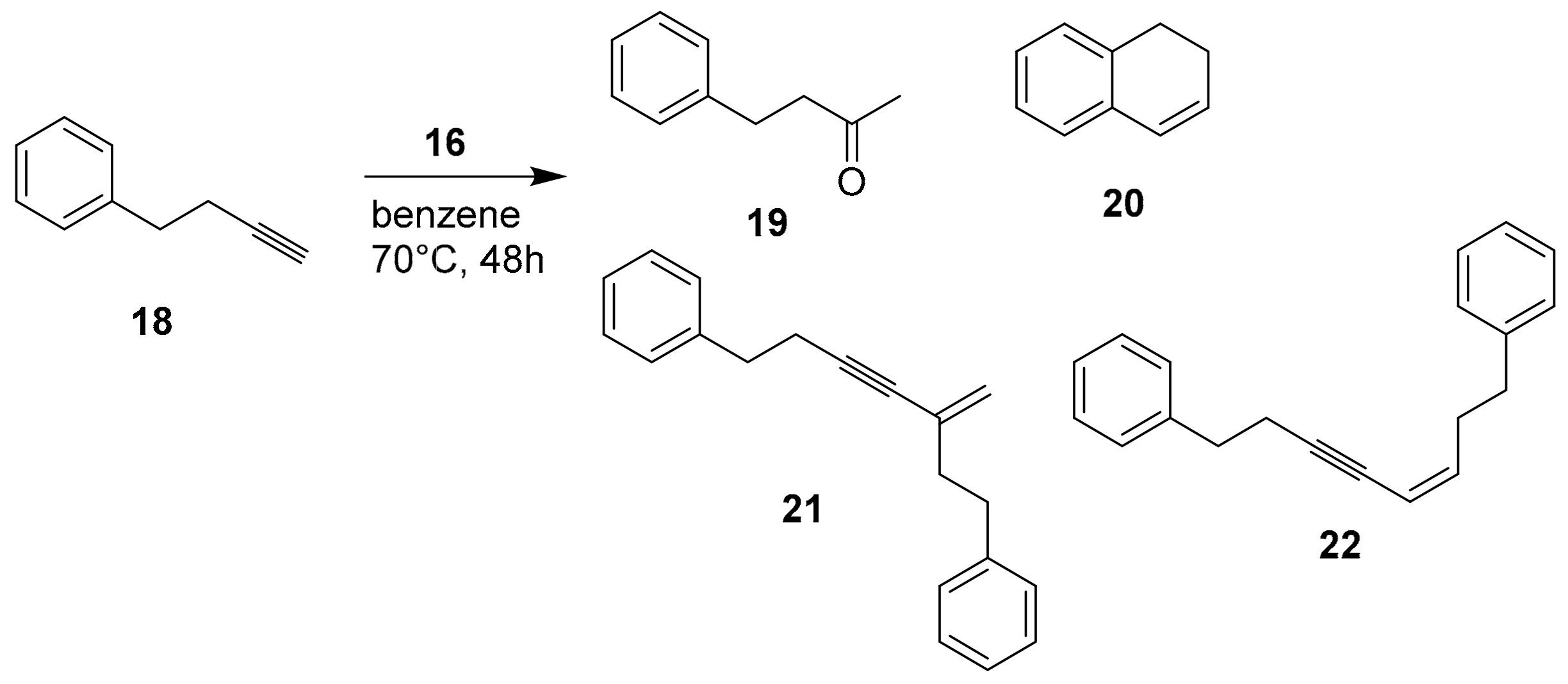
Furthermore, the authors tested also the change of the reactivity in the presence of capsule
2, using 4-phenyl-1-butyne
18 (
Scheme 7).
In the absence of the capsule, the catalyst can activate two molecules of alkyne, leading also to dimerization products
21 and
22 (
Table 5, entry 1). The capsule favors the intramolecularly cyclized product
20 (
Table 5, entry 2) due to the presence of mononuclear catalyst inside. The presence of the competitor (TEA) leads also to the dimerization products
21 and
22 (
Table 5, entry 3).
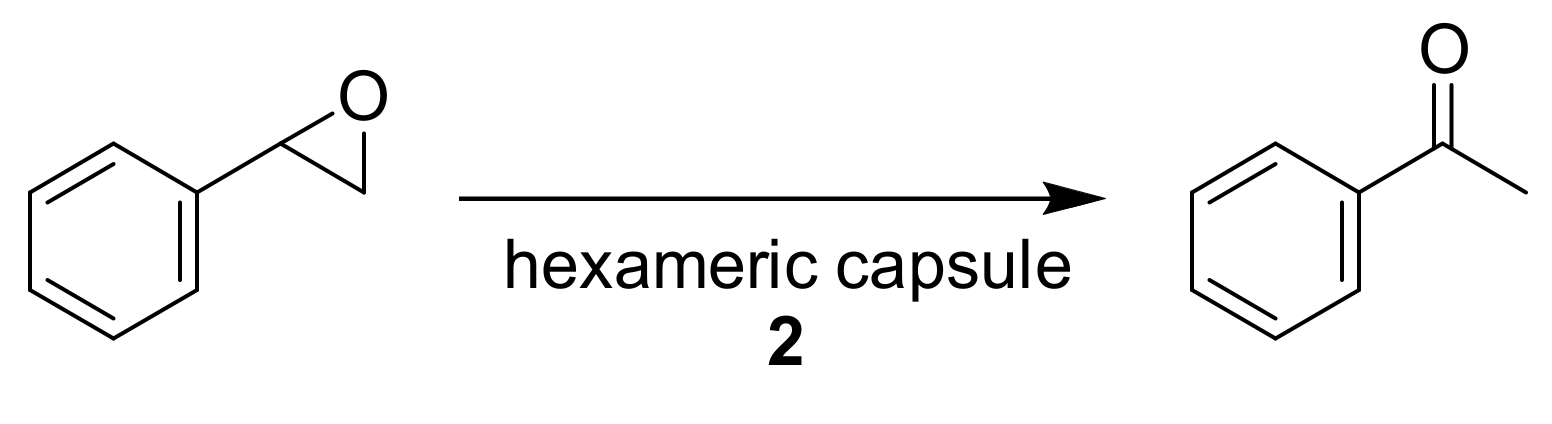
The metal-free isomerization of epoxides into the corresponding carbonyl compounds in a hexameric capsule was also tested by Scarso and coworkers (
Scheme 8) [
27]. The reaction was optimized by using styrene oxide as a substrate. In the absence of the self-assembled capsule, no isomerization product was observed. Only the presence of capsule
2 in catalytic amounts (7%) led to the formation of phenylacetaldehyde in good yield after 3 h (69%), while a quantitative yield was achieved after 18 h. This is a clear example of how the inner space of a supramolecular capsule can act as an enzyme, catalyzing reactions which do not occur in free solution.
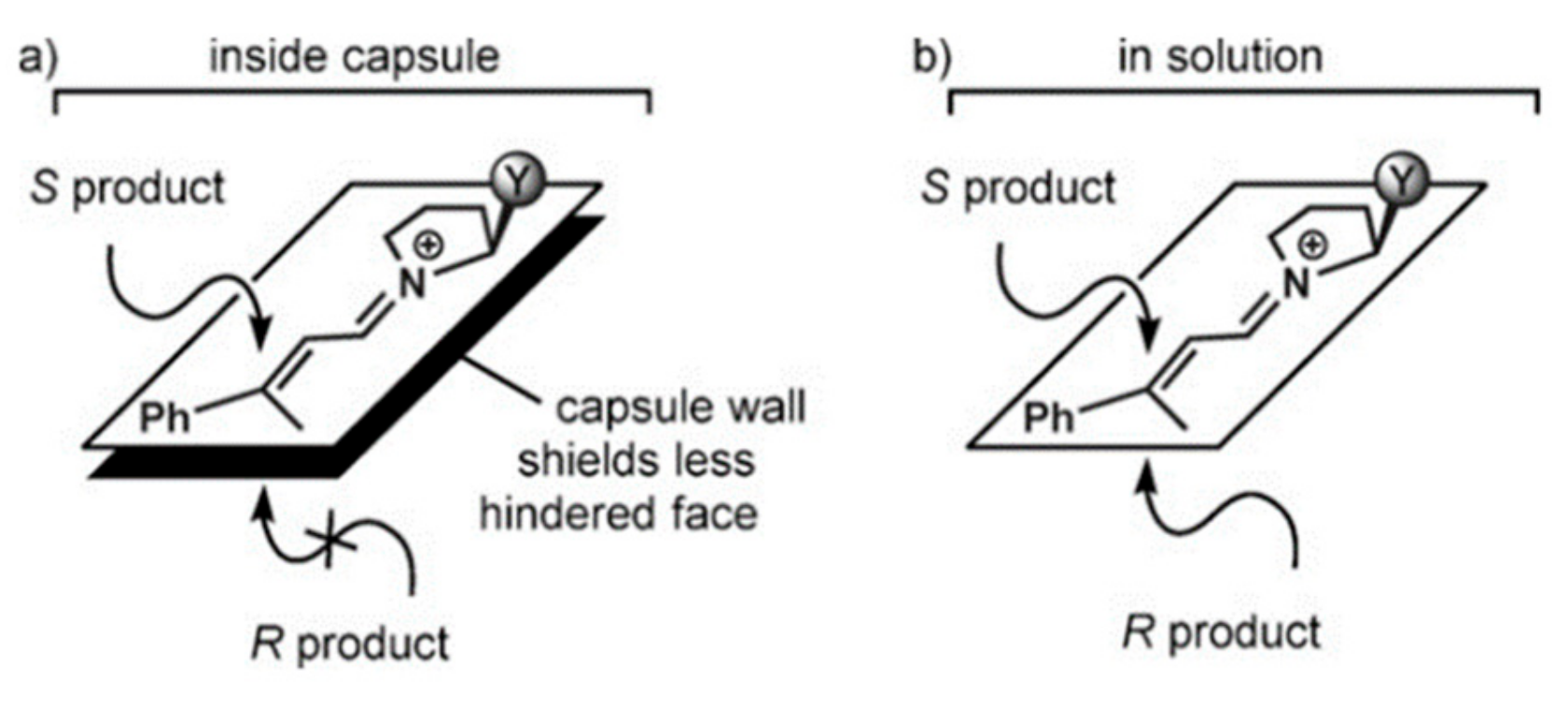
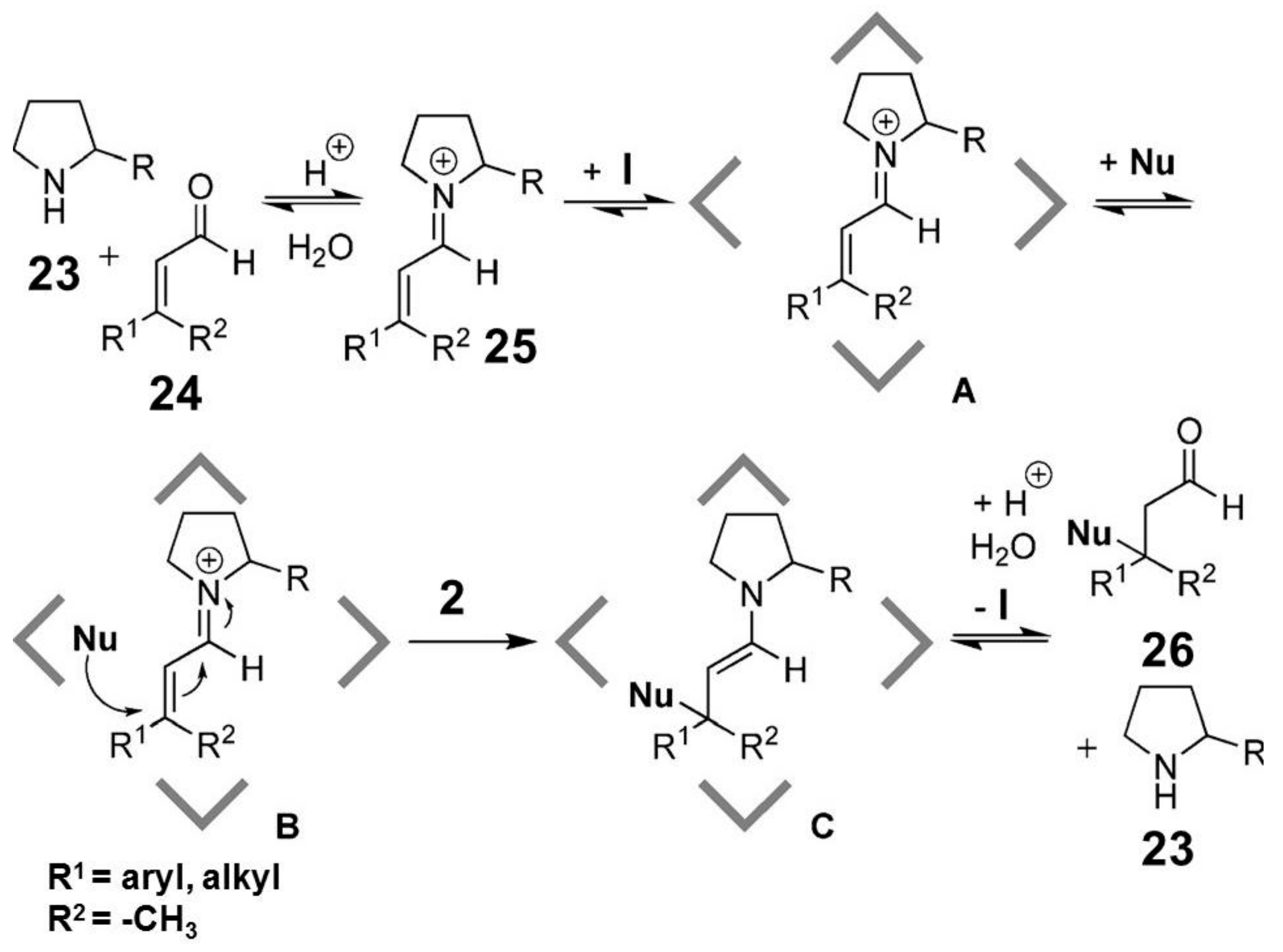
Tiefenbacher and coworkers tested a hexameric capsule as the nanoreactor for enantioselective catalysis by using a chiral iminium organocatalyst (
Scheme 9) [
28]. Recognition properties of the supramolecular capsule towards cationic species were exploited in order to include the reactive iminium species
25 inside the hydrophobic cavity, leading to intermediate
A. After the inclusion of the nucleophilic reagent (intermediate
B), reaction occurs inside the nanoreactor, leading to the product
26 and reforming the starting catalyst
23. By using chiral catalysts, the authors observed interesting results in term of enantiomeric excesses, observing reaction products with an
S-configuration. The authors proposed that iminium cation
25 interacts with the aromatic walls of the capsule via cation-π interactions from the less hindered face, selectively exposing the other face to the nucleophile (
Figure 3).
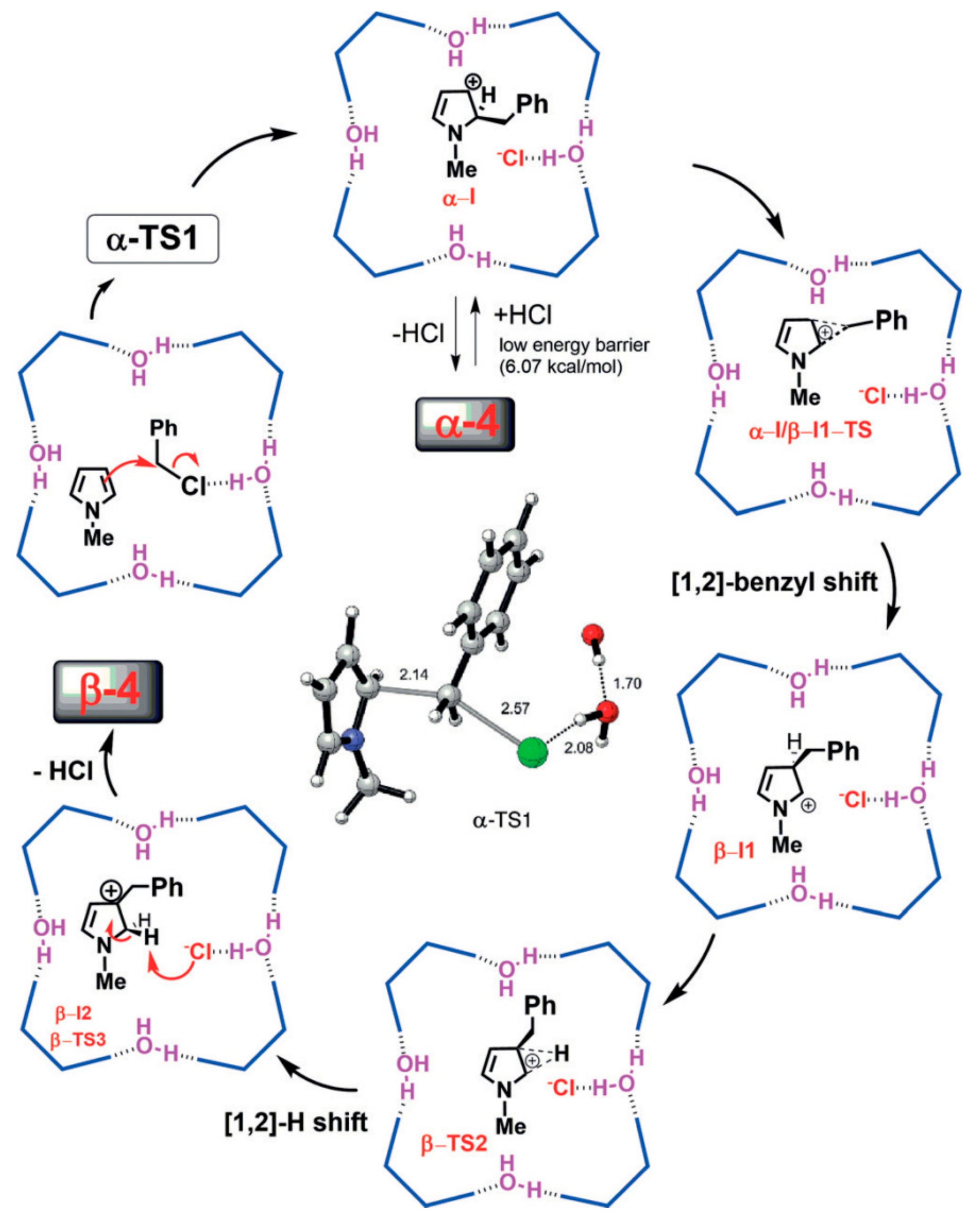
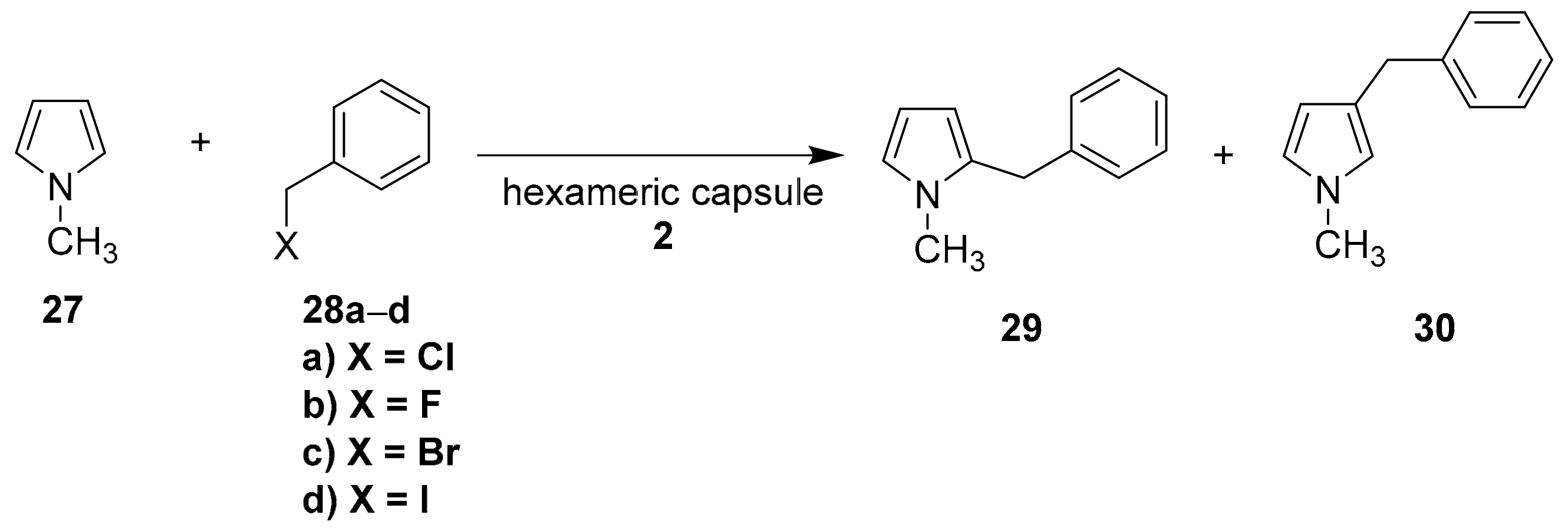
Recently, Neri and coworkers reported the use of the capsule
2 to catalyze the Friedel–Crafts benzylation of several arenes and heteroarenes with benzyl chloride under mild conditions [
29]. In particular, they tested the reaction between N-methylpyrrole
27 and benzyl halides (
28a–
d) in the presence of capsule
2 (
Scheme 10).
Reaction does not occur in the absence of the capsule (
Table 6, entry 1). In addition, the product of β-alkylation (
30) is favored in the presence of the capsule (
Table 6, entry 2), although in normal conditions, the preferred product is
29 [30]. These data demonstrate how the inner space of the capsule can change reactivity. By increasing the amount of capsule and the temperature, yields and
30/
29 ratio also increase (
Table 6, entry 4).
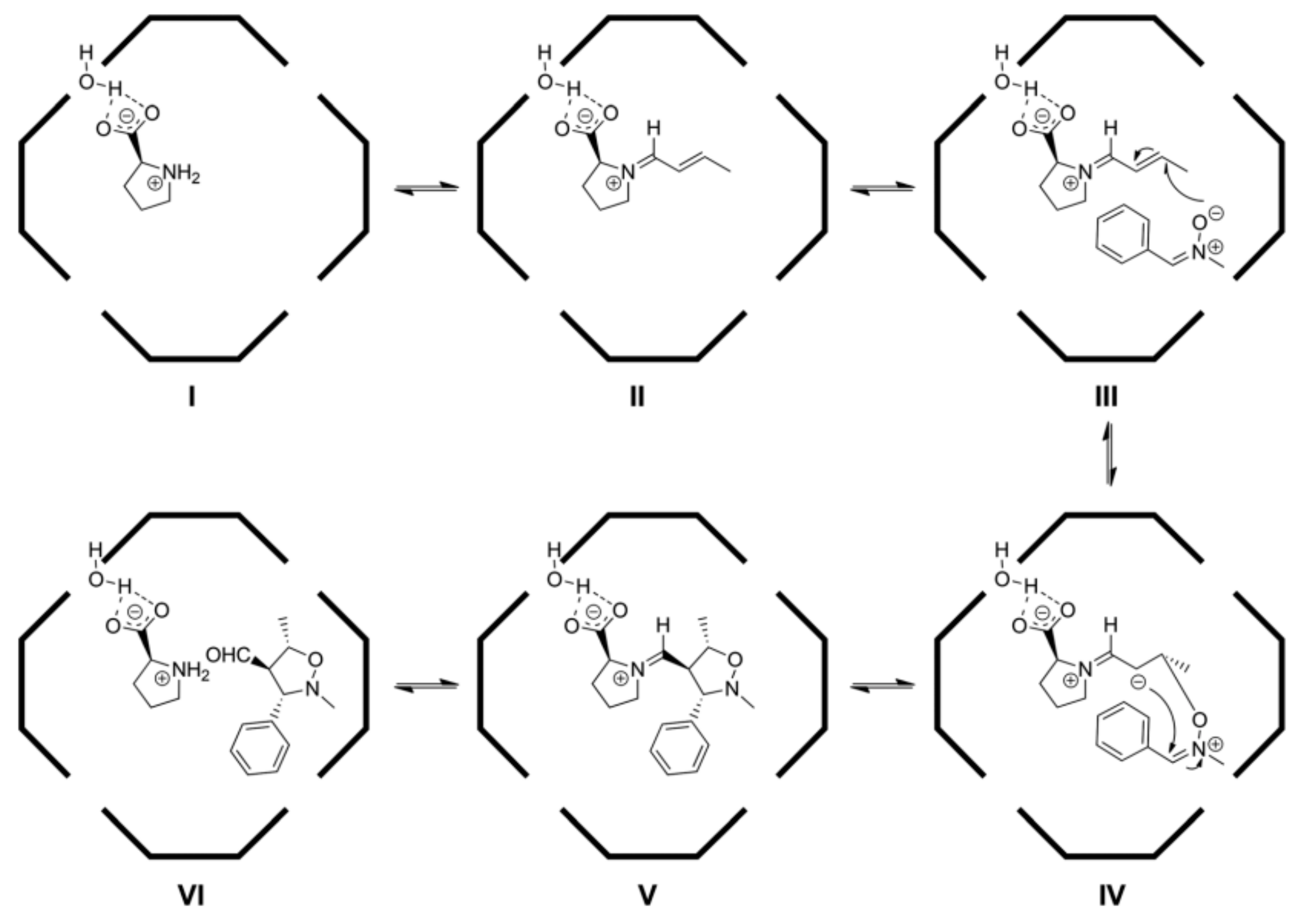
The authors performed quantum-chemical studies in order to understand the preference of β-alkylation inside the capsule (
Figure 4). In particular, some important pieces of evidence seem to justify the formation of
30: (
i) a hydrogen-bonding interaction between the chlorine atom and a bridging water molecule of the capsule; (
ii) two different p
Ka values (2.5 and 6.1) of hydrogen atoms directly bonded to oxygen atoms in the capsule; (
iii) the 1,2-benzyl shift.
The same research group employed self-assembled capsule
2 to perform 1,3-dipolar cycloaddition [
31]. In particular, they used nitrones and α, β-unsaturated aldehydes as substrates and
L-proline derivatives as catalysts (
Table 7). The presence of the capsule leads to a positive effect in the reaction. In all cases, in fact, the yields are higher with respect to the reaction conducted in the absence of the supramolecular cage. As reported in
Table 7, capsule
2 also influenced the stereochemistry of the reaction—in fact, the capsule leads to the formation of the
33-endo diastereoisomer. On the contrary, in free solution, the diastereoisomer
33-exo is favored. Furthermore, the enantioselectivity of the reaction is also strongly influenced by the hexameric capsule. Considering
33-endo, in the presence of the capsule, the enantiomer
4R is favored with respect to the
4S, while the trend is the opposite in the absence of capsule
2.
The authors performed a computer simulation to elucidate the mechanism involved (
Scheme 11). They suggested that the hexameric capsule, after inclusion of reactants, acts as acid catalyst for the iminium condensation.
3. Catalysis with Metallocage Capsules
Metal–ligand coordination shows an energy value of ca. 100–300 kJ/mol, and thus leads to the formation of a stable supramolecular capsule if the upper rim of a cavitand is functionalized with an appropriate chelating group able to bind a metal ion. After first pioneering study of Dalcanale and Jacopozzi [
32], many different metal-based supramolecular systems able to act as nanoreactors have been synthesized [
8,
9].
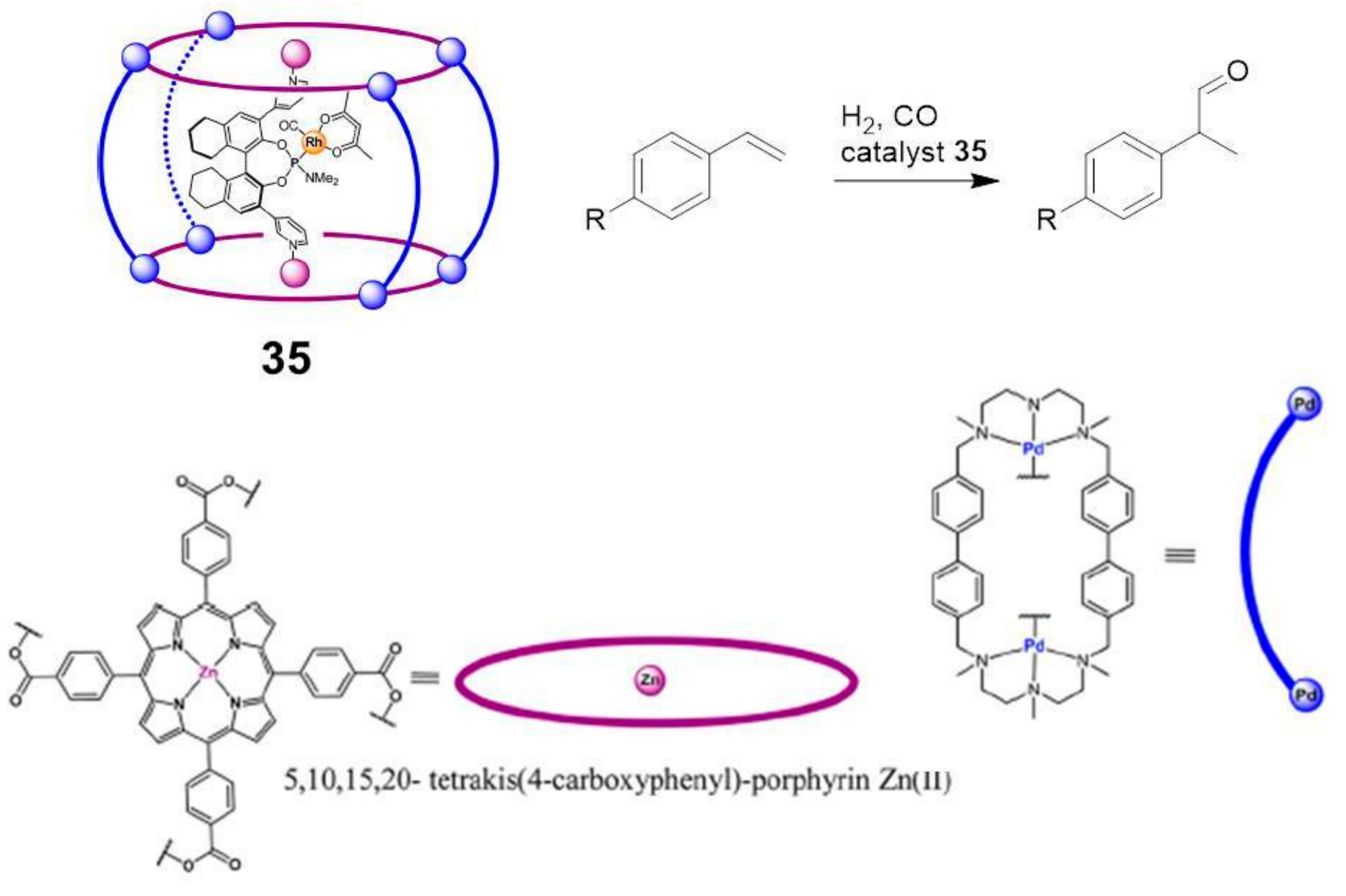
Recently, the research group of Prof. Reek synthesized a new tetragonal-prismatic nanocapsule,
35, containing two Zn-porphyrins linked by four Pd-based macrocyclic walls able to include a chiral Rh catalyst for the regio- and enantioselective hydroformylation of styrenes (
Figure 5) [
33].
In particular, substrates differed with respect to the R-substituents on the para position of the aromatic ring. By comparison with results obtained in bulk solution, the stereoselectivity observed with the cage is higher, due to the role of a second coordination sphere of the Rh catalyst.
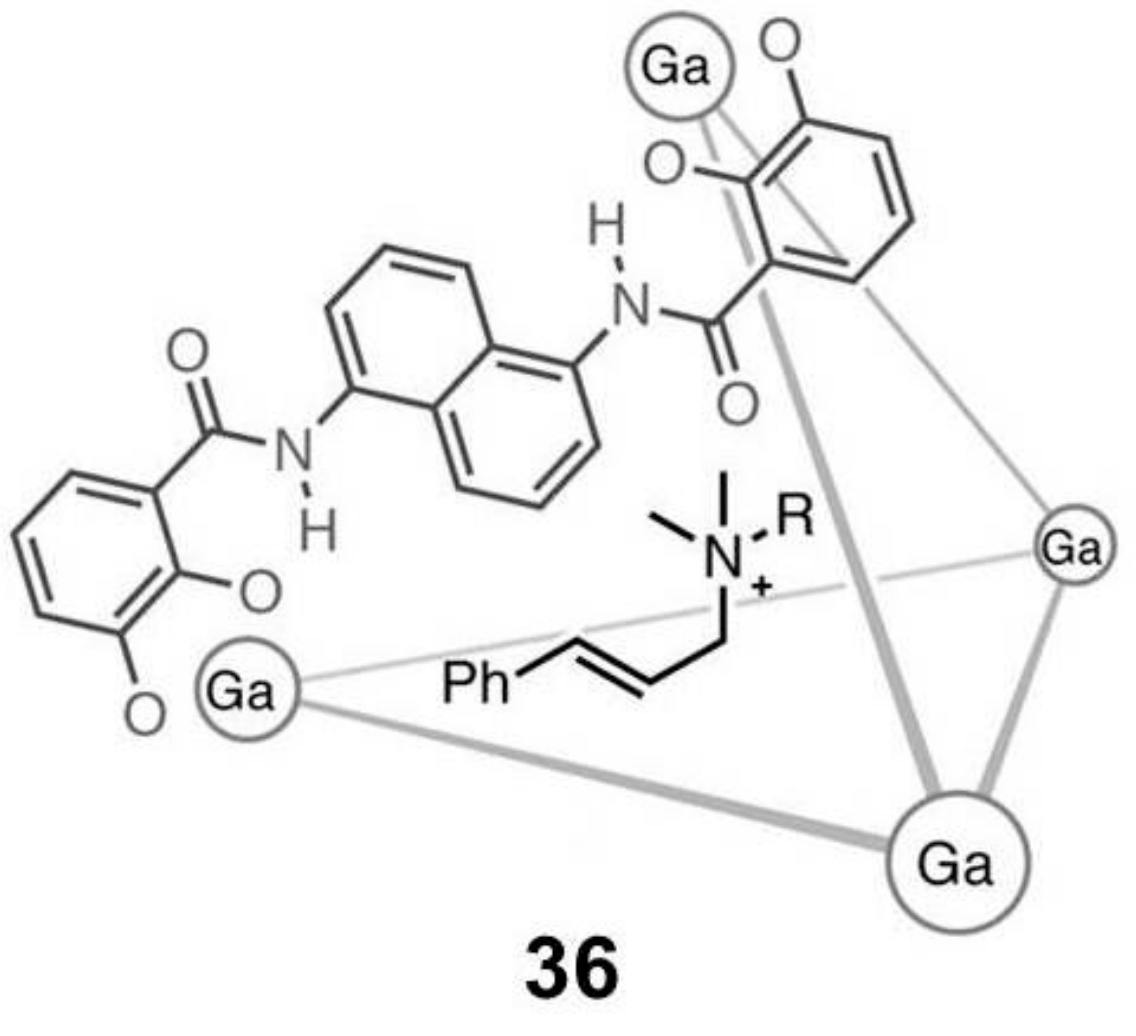
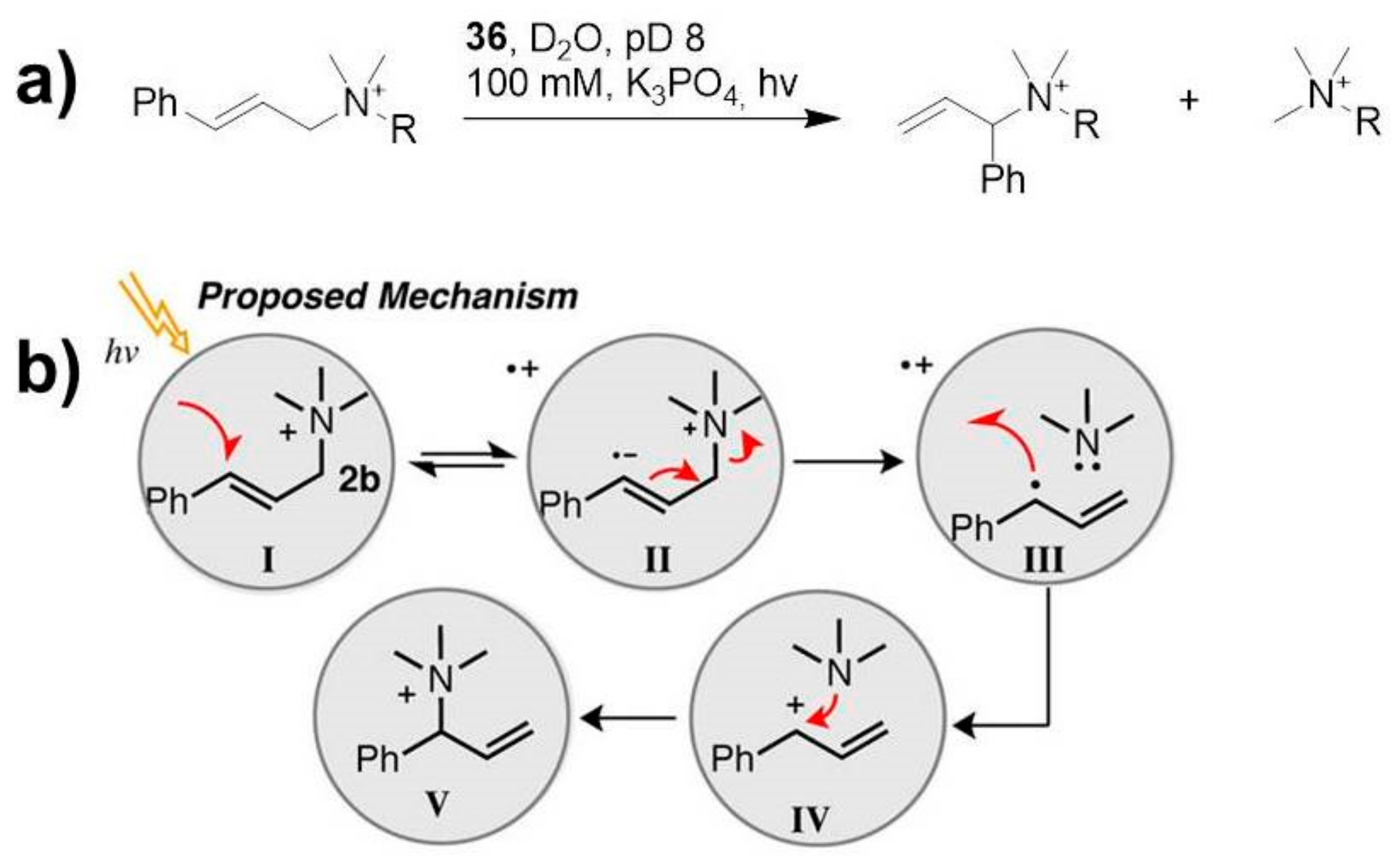
Raymond and coworkers reported the ability of a tetrahedral Ga-based capsule
36 (
Figure 6) to photosensitize the 1,3-rearrangement of cinnamylammonium cations from the linear to the higher-energy branched isomer after UVA irradiation [
34].
In particular, cage
36 acts as a photosensitizer, transferring the energy to the included guest, leading to a rearrangement not observed in solution (
Scheme 12).
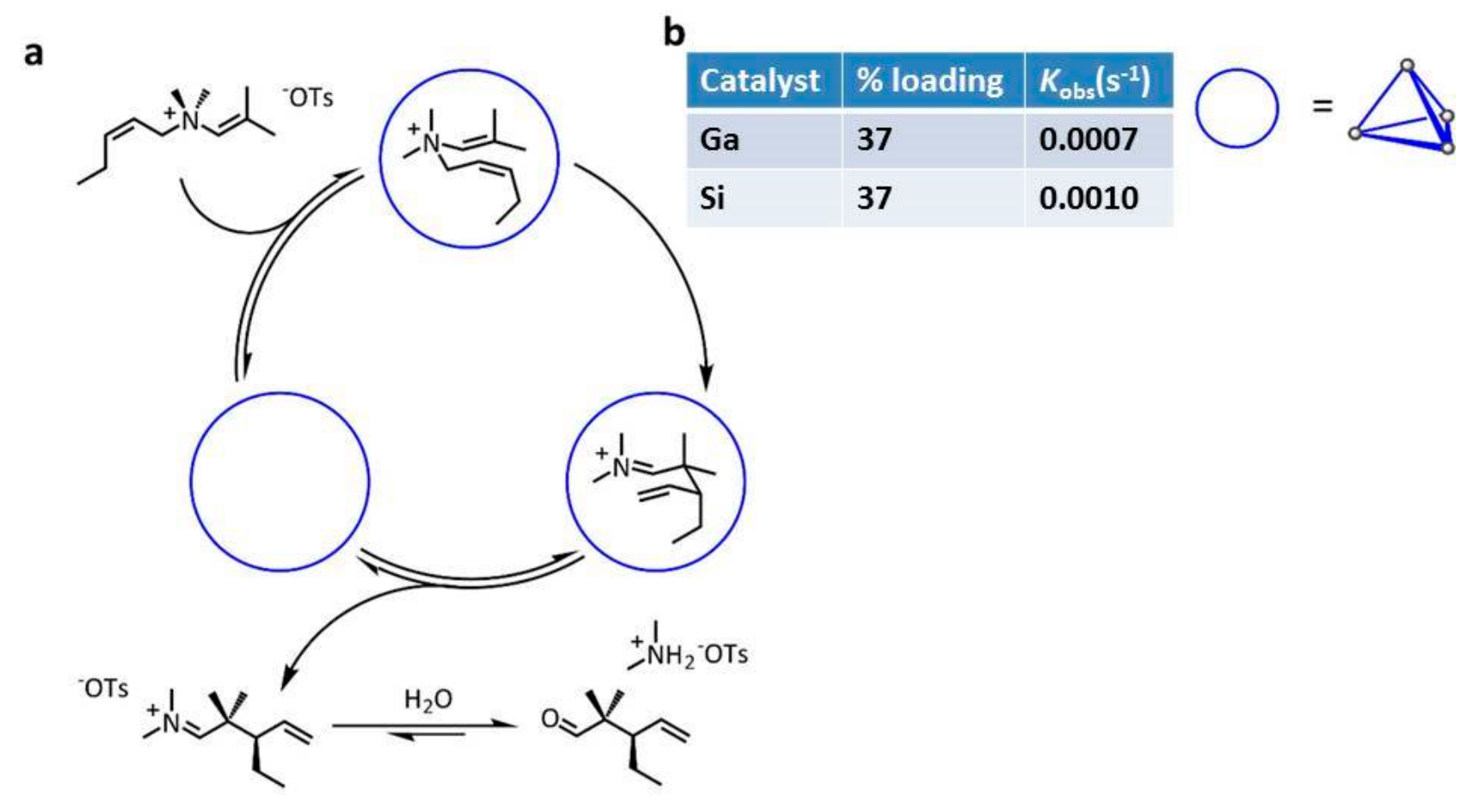
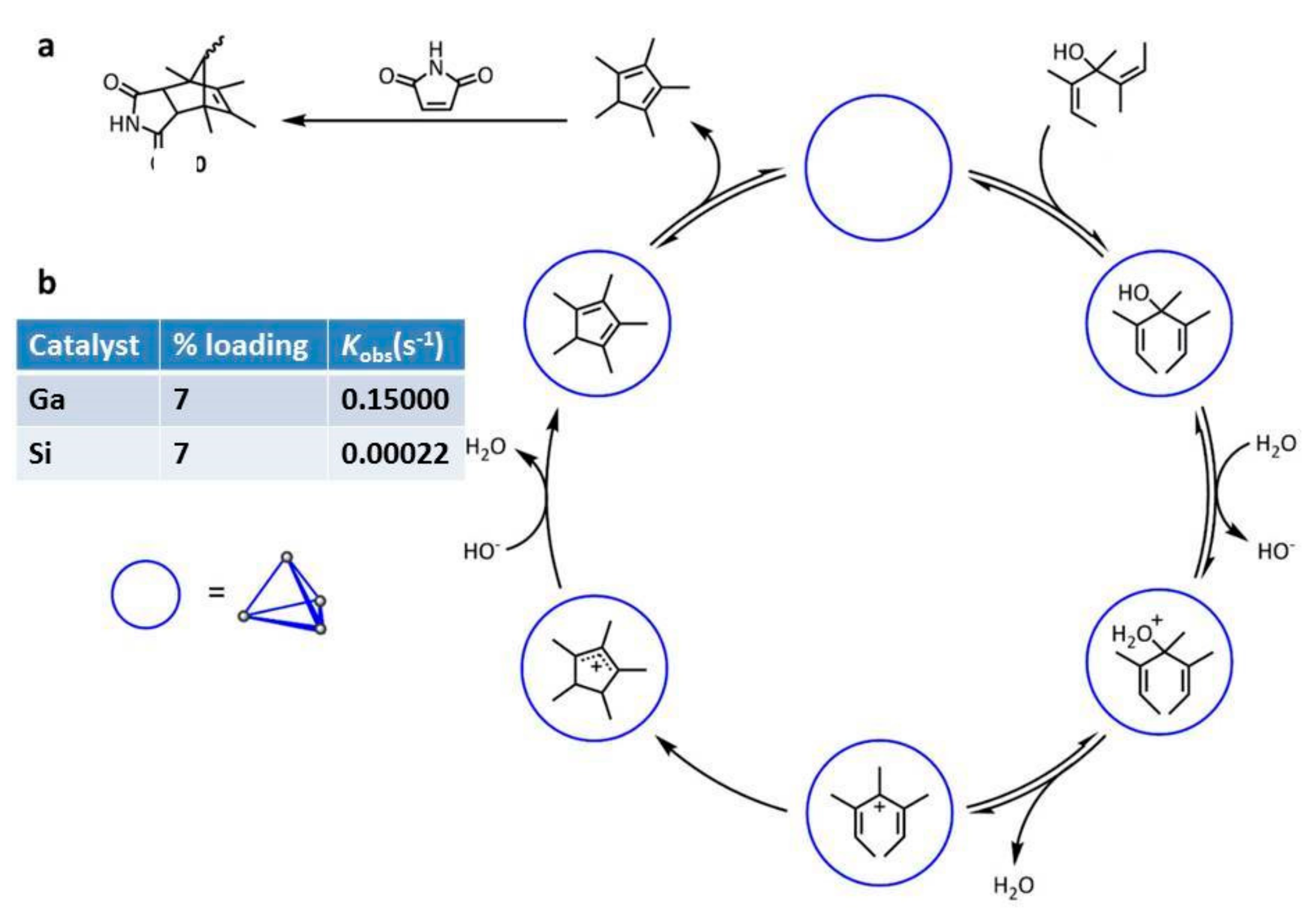
The same research group employed gadolinium capsule
36 and the analogue Si capsule in the aza-Cope rearrangement and in Nazarov cyclization (
Scheme 13 and
Scheme 14) [
35].
In particular, both metallocages show comparable catalytic activity for the aza-Cope rearrangement, while in the Nazarov rearrangement, a 680-fold increase of the reaction rate is observed. This is probably due to the increase of the charge of the starting neutral substrate, leading to cationic transition states stabilized by the anionic capsule.
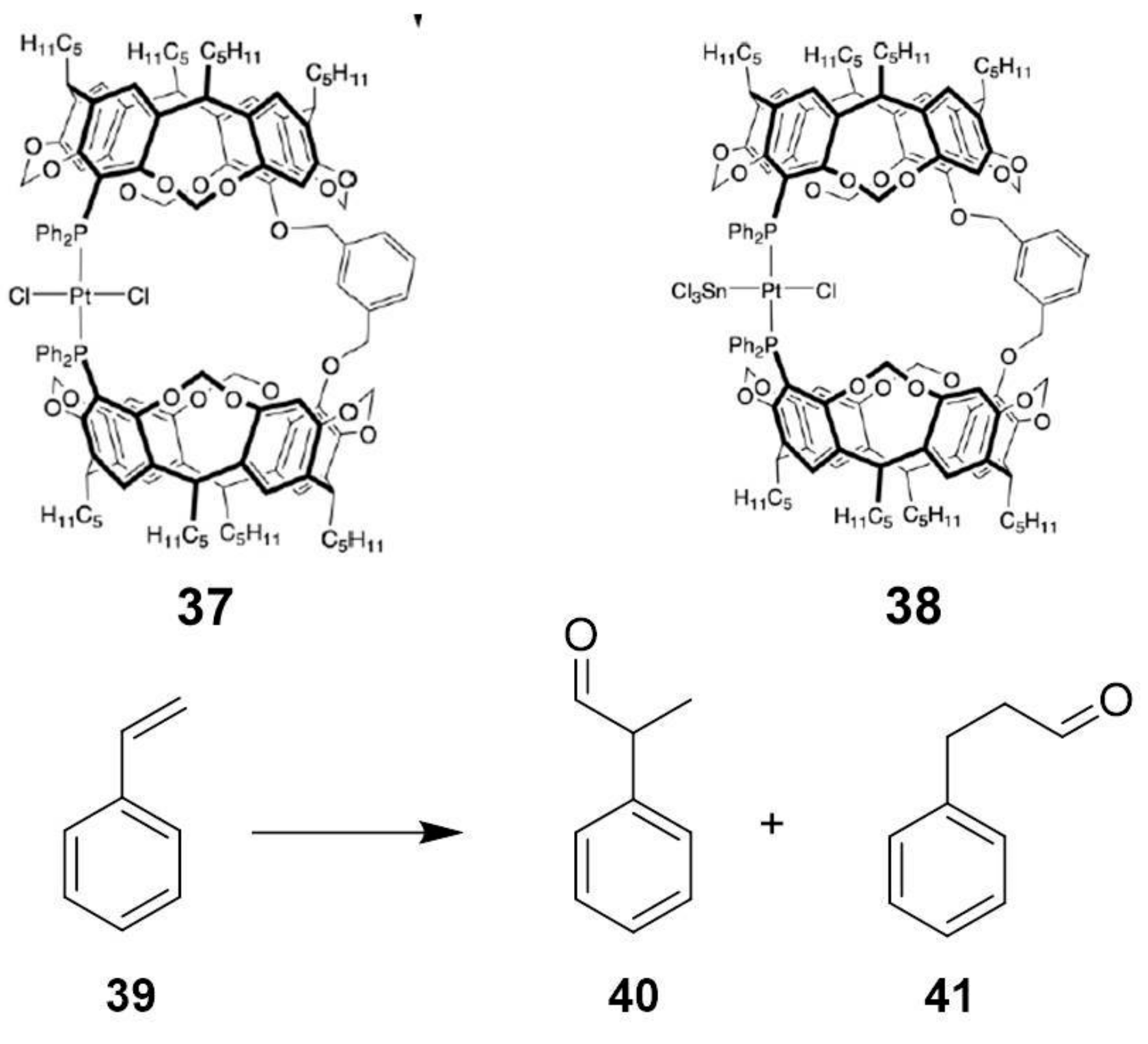
The research group of Prof. Matt employed a resorcinarene platform to build a metallocage able to catalyze the hydroformylation of styrene (
Scheme 15) [
36]. Due to a hydrophobic cavity having a diameter of ca. 8 Å, resorcinarene hosts possess an ideal platform to create nanoreactors [
37,
38]. In particular, two resorcinarene cavities can be linked with a phenyl ring and a Pt ion, leading to nanocages
37 and
38 (
Scheme 15).
Table 8 reports the results of the hydroformylation of styrene obtained by using metallocages
37 and
38. While the noncapsular catalyst leads to similar amounts of branched and linear products (
40 and
41, respectively) in 24 h, the nanocages lead to the preferential linear aldehyde
41. Molecular modeling suggested that steric interactions between the guest and the capsule walls are slightly lower for the branched guest than for the linear one, thus explaining the observed selectivity.
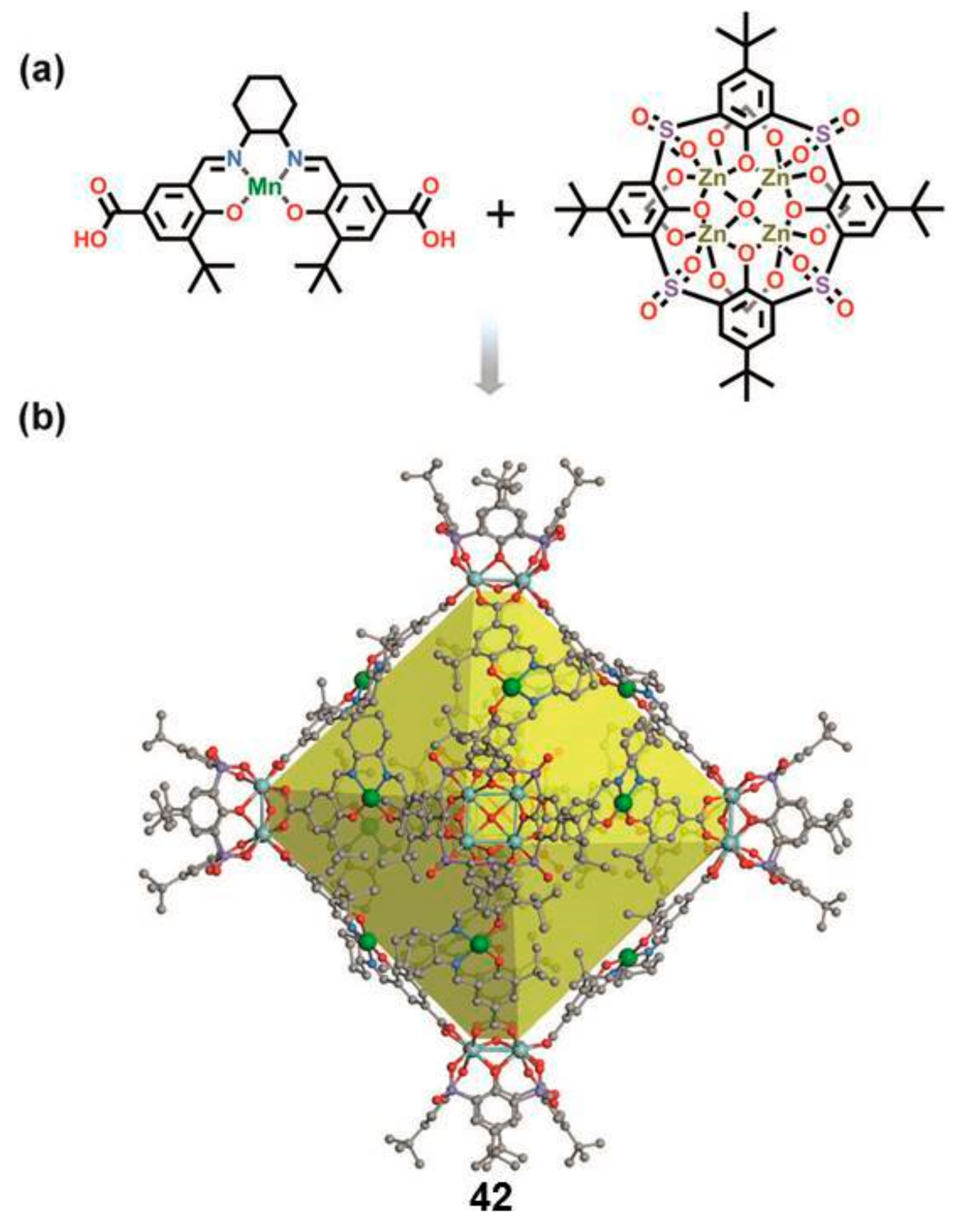
Liu and Cui reported on the supramolecular container
42, constructed by using 12 enantiopure Mn(salen)-derived dicarboxylic acids as linear linkers and six Zn4-
p-
tert-butylsulfonylcalix [
4] arene clusters as tetravalent four-connected vertices (
Figure 7), leading to a chiral octahedral coordination cage that can act as a supramolecular catalyst for the oxidative kinetic resolution (OKR) of racemic alcohols and asymmetric epoxidation of olefins with higher reactivity/enantioselectivity compared with the free metallosalen catalyst [
39].
The cage is assembled by the reaction of sulfonylcalix [
4] arenes with zinc ions to furnish a shuttlecock-like tetrametallic cluster, which is known to be a four-connected node in making cages, exploiting the coordination with supplementary organic linkers [
40].
The authors firstly demonstrated the catalytic activity of the chiral cage in the OKR of racemic secondary alcohols to afford enantiomerically pure secondary alcohols, which are often employed as starting materials for pharmaceutical compounds. All the reactions showed good conversions and excellent enantioselectivities (54–59% conversion and 91–99% of enantiomeric excess) for 1-phenylethanol and its derivatives possessing both electron-withdrawing and electron-donating substituents on the meta/para-position, while ortho-substituted 1-phenylethanol derivatives offered no or low ee values. The resolution of 1-phenylethanol by the chiral supramolecular catalyst afforded the R enantiomer in preference to the S enantiomer (96% ee), indicating that the enantioselectivity of the reaction is controlled by the handedness of the cage. Furthermore, the catalytic reaction with 1-phenyl-2-propanol, 1-indanol, and their derivatives led to ee values of 86–99.3%. The catalytic activity of the chiral cage was compared with the Mn–Jacobsen catalyst (a well-known catalyst for asymmetric oxidation reactions) for the OKR of 1-indanol, establishing that when the loading was reduced from 0.25 to 0.10 mol %, the chiral cage was still able to afford 60% ee of the product, while the Jacobsen catalyst led to a reaction with no or very little enantioselectivity.
The chiral supramolecular cage was also used as a supramolecular catalyst for epoxidation reactions of 2,2-dimethyl-2H-chromene and its derivatives. All the epoxidation reactions were performed with 0.05 mol % of catalyst with 2-(tert-butylsulfonyl) iodosylbenzene (sPhIO) as an oxidant in CH2Cl2 at 0 °C, affording the epoxides in 81–96% ee. These results highlight that the cage architecture is able to stabilize the Mn(salen) catalysts, avoid intermolecular deactivation, and encapsulate substrates and reactants in the cavity, increasing reactivity and enantioselectivity.
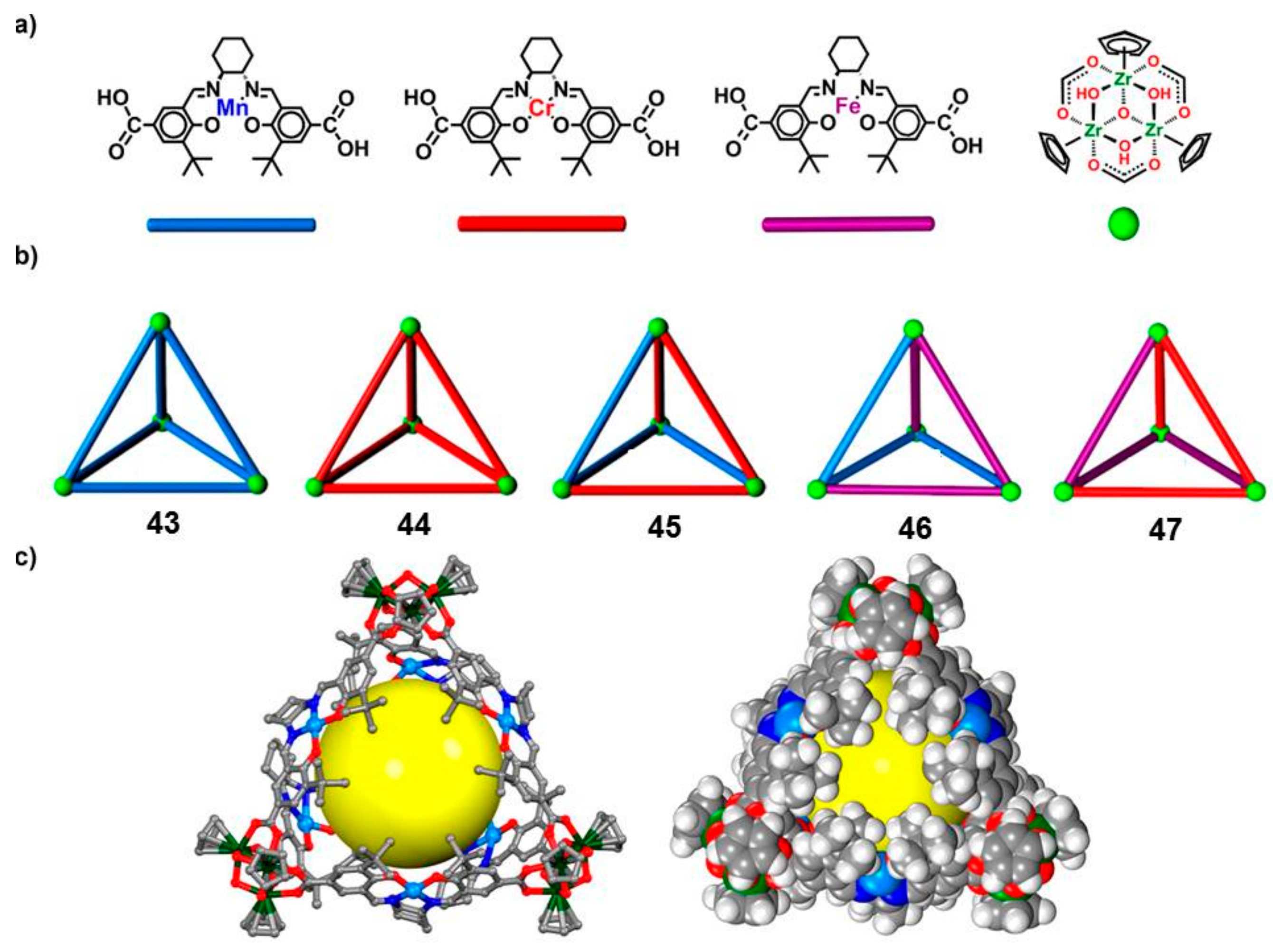
Cui and coworkers reported on the assembly of five chiral single- and mixed-linker tetrahedral coordination cages (
43–
47,
Figure 8) using six enantiopure Mn, Cr, and/or Fe(salen) complexes as linear linkers and four Cp
3Zr
3 clusters as three-connected vertices [
41].
It is well known that Mn(salen) and Fe(salen) derivatives are excellent catalysts for the enantioselective epoxidation of alkenes [
42,
43,
44,
45,
46], whereas the Cr(salen) complexes are catalysts for epoxide ring-opening reactions. These supramolecular nanoreactors feature a nanoscale hydrophobic cavity bearing the same or different catalytically active sites. In particular, the cage constructed with Mn(salen) complexes was able to catalyze the asymmetric epoxidation of alkenes, while the cage bearing Cr(salen) complexes catalyzed the ring opening of epoxides. Prompted by these results, the authors assembled a chiral cage bearing both Mn and Cr salen derivatives in which the presence of two different catalytic centers promotes sequential reactions initiated by the epoxidation of alkenes, followed by the ring opening of epoxides. The study of the sequential reaction was initiated with exposure of 2,2-dimethyl-2H-chromene (DMCH) to PhIO ((diacetoxyiodo) benzene) and a Mn–Cr cage to afford the corresponding epoxide, followed by addiction of trimethylsilylazide (TMSN
3) to support the ring opening of the epoxide, providing the azido alcohol in 85% DMCH conversion and 79% yield with 93% ee. Also, DMCH derivatives bearing methyl and nitro groups afforded 78–88% alkene conversions and 73–81% yields with up to 93% ee.
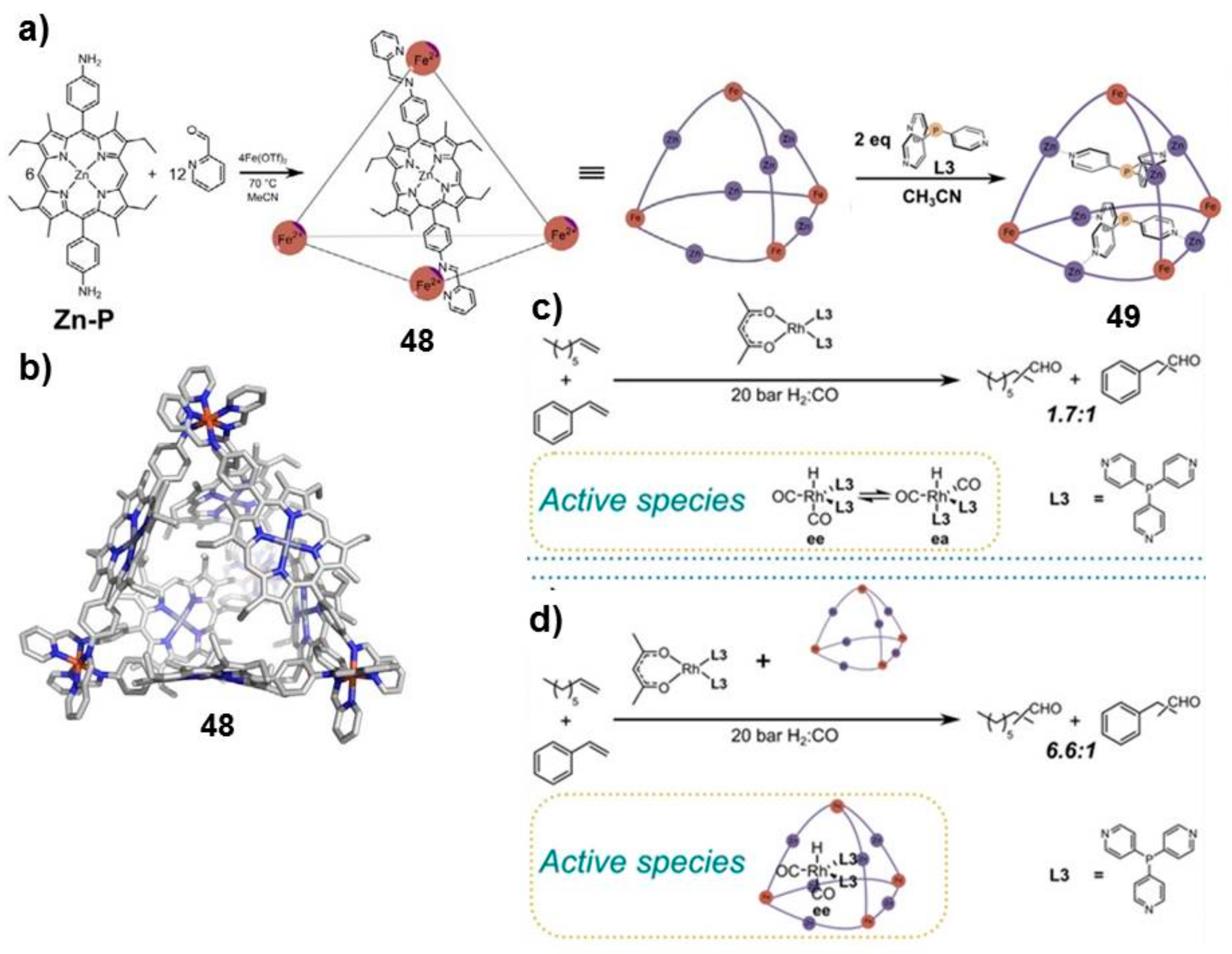
Reek and coworkers reported the first example of the size-selective hydroformylation of alkenes by encapsulation of a rhodium phosphine complex in the molecular cage
48 (
Figure 9) [
47]. The rhodium complex is strongly bound inside the cavity by a ligand-template approach, due to the presence of pyridyl–zinc (II) porphyrins that led to high association constants (>10
5 M
−1). DFT calculations and in situ high-pressure IR studies of the host–guest complex corroborate the evidence that the encapsulated active species adopt the ee coordination geometry, in which both phosphine ligands are in equatorial positions.
Upon binding of rhodium catalyst, the inner space and the window aperture of the cage slightly decrease, leading to a slower diffusion of larger substrates into the cage compared to that of smaller substrates, which are easily transformed into the corresponding aldehydes. Notably, after activation of the catalyst precursor, followed by the addition of the cage
48, 1-octene was converted into the corresponding aldehyde with 78% conversion in 72 h at 70 °C, whereas for both selected aromatic guests (styrene and 4-
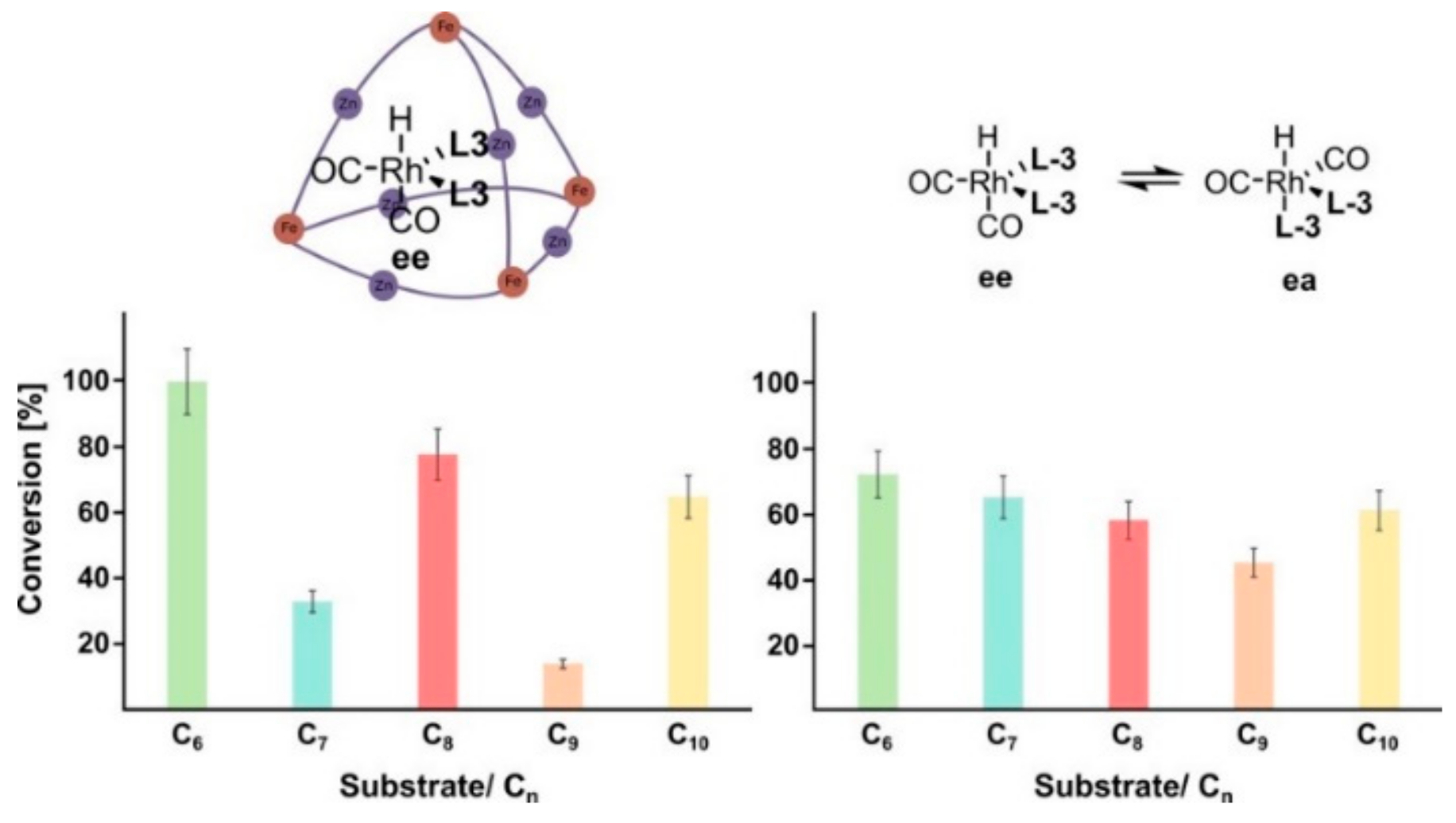
tert-butylstyrene), 0% conversion was observed, thus confirming that the encapsulation of rhodium catalyst prevents the access of bulky guests inside the cavity. Furthermore, the authors investigated the reaction of a series of aliphatic alkenes (from 1-hexene to 1-decene; see
Figure 10), highlighting that the catalyst preferentially converted shorter substrates.
From these data, an odd–even effect is also evident. This particular effect probably depends on an enthalpically more favorable folding of some alkenes inside the cavity. Such an effect is not seen in solution, where the nonencapsulated catalyst transforms all substrates with comparable conversion values.































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





