Calculation of Surface Properties of Cubic and Hexagonal Crystals through Molecular Statics Simulations

Abstract
:1. Introduction
2. Methodology
2.1. Bulk Elastic Constants
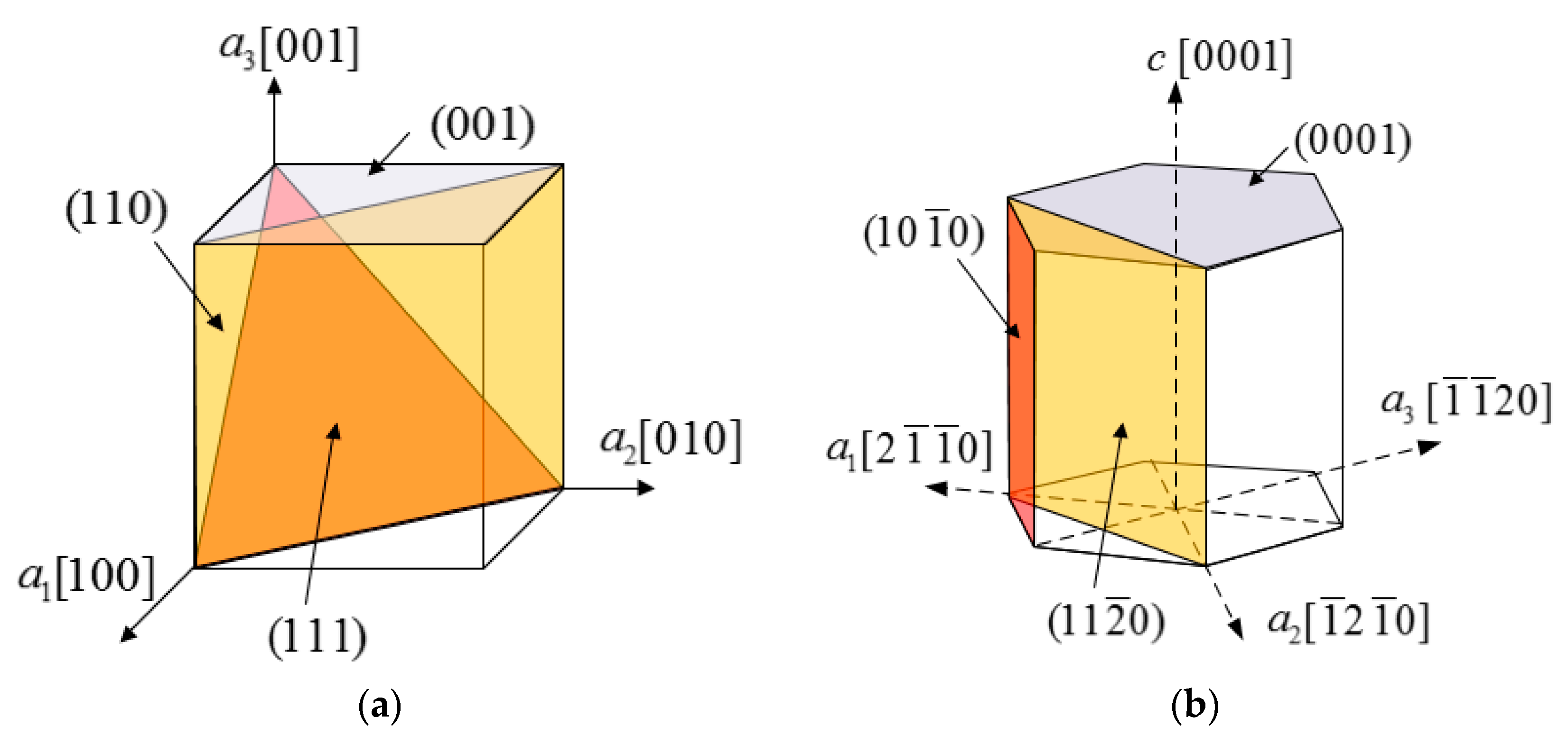
2.2. Surface Properties
3. Results
3.1. Bulk Elastic Constants
3.2. Surface Properties
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ye, W.; Chen, B. Elastic relaxation in 3D epitaxial nanoisland with strain-dependent surface stress effect. J. Cryst. Growth 2015, 410, 59–62. [Google Scholar] [CrossRef]
- Bickel, J.E.; Millunchick, J.M. The impact of the initial surface reconstruction on heteroepitaxial film growth and defect formation. Phys. Scr. 2014, 89. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, C.E.; Janotti, A.; Van de Walle, C.G. Absolute surface energies of polar and nonpolar planes of GaN. Phys. Rev. B 2014, 89. [Google Scholar] [CrossRef]
- Diao, Y.; Liu, L.; Xia, S.H. Adsorption mechanism of Pt, Ag, Al, Au on GaAs nanowire surfaces from first-principles. J. Phys. Condens. Matter 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Liu, Y. Elastic and piezoelectric fields around a quantum wire of zincblende heterostructures with interface elasticity effect. Appl. Phys. A 2018, 124, 285. [Google Scholar] [CrossRef]
- Fang, X.-Q.; Zhu, C.-S.; Liu, J.-X.; Liu, X.-L. Surface energy effect on free vibration of nano-sized piezoelectric double-shell structures. Phys. B-Condens. Matter 2018, 529, 41–56. [Google Scholar] [CrossRef]
- Zhang, S.; Tang, X.; Ruan, H.; Zhu, L. Effects of surface/interface stress on phonon properties and thermal conductivity in AlN/GaN/AlN heterostructural nanofilms. Appl. Phys. A-Mater. Sci. Process. 2019, 125. [Google Scholar] [CrossRef]
- Gao, Z.; Lu, L.; Xue, X.; Li, J.; Zhao, L.; Ahmad, D.; Li, H. Comparative Study of ZnO Nanostructures Grown on Variously Orientated GaN and AlxGa1-xN: The Role of Polarization, and Surface Pits. Crystals 2019, 9, 663. [Google Scholar] [CrossRef]
- Resel, R.; Koini, M.; Novak, J.; Berkebile, S.; Koller, G.; Ramsey, M. Epitaxial Order Driven by Surface Corrugation: Quinquephenyl Crystals on a Cu(110)-(2x1)O Surface. Crystals 2019, 9, 373. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yan, H.; Zhu, G.; Liu, S.; Gan, Z.; Zhang, Z. Effect of Substrate Surface on Deposition of AlGaN: A Molecular Dynamics Simulation. Crystals 2018, 8, 279. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, X.R.; Beom, H.G. Effect of Side Surface Orientation on the Mechanical Properties of Silicon Nanowires: A Molecular Dynamics Study. Crystals 2019, 9, 102. [Google Scholar] [CrossRef] [Green Version]
- Cammarata, R.C. Surface and interface stress effects in thin films. Prog. Surf. Sci. 1994, 46, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Shuttleworth, R. The surface tension of solids. Proc. Phys. Soc. Sect. A 1950, 63, 444. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals. Phys. Rev. B 1984, 29, 6443–6453. [Google Scholar] [CrossRef] [Green Version]
- Foiles, S.M.; Baskes, M.I.; Daw, M.S. Embedded-atom-method functions for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys. Phys. Rev. B 1986, 33, 7983–7991. [Google Scholar] [CrossRef] [PubMed]
- Finnis, M.W.; Sinclair, J.E. A simple empirical N-body potential for transition metals. Philos. Mag. A 1984, 50, 45–55. [Google Scholar] [CrossRef]
- Gumbsch, P.; Daw, M.S. Interface stresses and their effects on the elastic moduli of metallic multilayers. Phys. Rev. B 1991, 44, 3934–3938. [Google Scholar] [CrossRef]
- Feibelman, P.J. Anisotropy of the stress on fcc(110) surfaces. Phys. Rev. B 1995, 51, 17867–17875. [Google Scholar] [CrossRef]
- Ackland, G.J.; Finnis, M.W. Semi-empirical calculation of solid surface tensions in body-centred cubic transition metals. Philos. Mag. A 1986, 54, 301–315. [Google Scholar] [CrossRef]
- Ackland, G.J.; Thetford, R. An improved N-body semi-empirical model for body-centred cubic transition metals. Philos. Mag. A 1987, 56, 15–30. [Google Scholar] [CrossRef]
- Ackland, G.J. Theoretical study of titanium surfaces and defects with a new many-body potential. Philos. Mag. A 1992, 66, 917–932. [Google Scholar] [CrossRef] [Green Version]
- Ackland, G.J.; Wooding, S.J.; Bacon, D.J. Defect, surface and displacement-threshold properties of α-zirconium simulated with a many-body potential. Philos. Mag. A 1995, 71, 553–565. [Google Scholar] [CrossRef]
- Northrup, J.E. Dimer-plus-chain structure for the Si(100)-c(4×2) surface. Phys. Rev. Lett. 1985, 54, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Northrup, J.E.; Cohen, M.L. Reconstruction Mechanism and Surface-State Dispersion for Si(111)-(2×1). Phys. Rev. Lett. 1982, 49, 1349–1352. [Google Scholar] [CrossRef]
- Meade, R.D.; Vanderbilt, D. Adatoms on Si(111) and Ge(111) surfaces. Phys. Rev. B 1989, 40, 3905–3913. [Google Scholar] [CrossRef]
- Meade, R.D.; Vanderbilt, D. Origins of stress on elemental and chemisorbed semiconductor surfaces. Phys. Rev. Lett. 1989, 63, 1404–1407. [Google Scholar] [CrossRef]
- Kaxiras, E.; Bar-Yam, Y.; Joannopoulos, J.D.; Pandey, K.C. (2×2) reconstructions of the {111} polar surfaces of GaAs. Phys. Rev. B 1986, 33, 4406–4409. [Google Scholar] [CrossRef] [Green Version]
- Qian, G.-X.; Martin, R.M.; Chadi, D.J. First-principles calculations of atomic and electronic structure of the GaAs(110) surface. Phys. Rev. B 1988, 37, 1303–1307. [Google Scholar] [CrossRef]
- Qian, G.-X.; Martin, R.M.; Chadi, D.J. First-principles study of the atomic reconstructions and energies of Ga- and As-stabilized GaAs(100) surfaces. Phys. Rev. B 1988, 38, 7649–7663. [Google Scholar] [CrossRef]
- Northrup, J.E.; Neugebauer, J. Indium-induced changes in GaN(0001) surface morphology. Phys. Rev. B 1999, 60, R8473–R8476. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadeh-Bakhshayesh, H.; Gutkin, M.Y.; Shodja, H.M. Surface/interface effects on elastic behavior of a screw dislocation in an eccentric core-shell nanowire. Int. J. Solids Struct. 2012, 49, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Fang, Q.H.; Liu, Y.W. Size-dependent elastic interaction of a screw dislocation with a circular nano-inhomogeneity incorporating interface stress. Scr. Mater. 2006, 55, 99–102. [Google Scholar] [CrossRef]
- Fang, Q.H.; Liu, Y.W.; Jin, B.; Wen, P.H. Interaction between a dislocation and a core-shell nanowire with interface effects. Int. J. Solids Struct. 2009, 46, 1539–1546. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ye, W. Elastic and piezoelectric field around a quantum dot with interface effect. Phys. E: Low-Dimens. Syst. Nanostruct. 2017, 89, 5–9. [Google Scholar] [CrossRef]
- Müller, P. Elastic effects on surface physics. Surf. Sci. Rep. 2004, 54, 157–258. [Google Scholar] [CrossRef]
- Shodja, H.M.; Gutkin, M.Y.; Moeini-Ardakani, S.S. Effect of surface stresses on elastic behavior of a screw dislocation inside the wall of a nanotube. Phys. Status Slidi 2011, 248, 1437–1441. [Google Scholar] [CrossRef]
- Miller, R.E.; Shenoy, V.B. Size-dependent elastic properties of nanosized structural elements. Nanotechnology 2000, 11, 139. [Google Scholar] [CrossRef]
- Dingreville, R.; Qu, J.M.; Cherkaoui, M. Surface free energy and its effect on the elastic behavior of nano-sized particles, wires and films. J. Mech. Phys. Solids 2005, 53, 1827–1854. [Google Scholar] [CrossRef]
- Shenoy, V.B. Atomistic calculations of elastic properties of metallic fcc crystal surfaces. Phys. Rev. B 2005, 71, 094104–094111. [Google Scholar] [CrossRef] [Green Version]
- Dingreville, R.; Qu, J. A semi-analytical method to compute surface elastic properties. Acta Mater. 2007, 55, 141–147. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Mishin, Y.; Asta, M.; Li, J. Atomistic modeling of interfaces and their impact on microstructure and properties. Acta Mater. 2010, 58, 1117–1151. [Google Scholar] [CrossRef] [Green Version]
- Cowley, E.R. Lattice Dynamics of Silicon with Empirical Many-Body Potentials. Phys. Rev. Lett. 1988, 60, 2379–2381. [Google Scholar] [CrossRef] [PubMed]
- Albe, K.; Nordlund, K.; Nord, J.; Kuronen, A. Modeling of compound semiconductors: Analytical bond-order potential for Ga, As, and GaAs. Phys. Rev. B 2002, 66. [Google Scholar] [CrossRef] [Green Version]
- Nord, J.; Albe, K.; Erhart, P.; Nordlund, K. Modelling of compound semiconductors: Analytical bond-order potential for gallium, nitrogen and gallium nitride. J. Phys. Condens. Matter 2003, 15, 5649. [Google Scholar] [CrossRef]
- Sun, D.Y.; Mendelev, M.I.; Becker, C.A.; Kudin, K.; Haxhimali, T.; Asta, M.; Hoyt, J.J.; Karma, A.; Srolovitz, D.J. Crystal-melt interfacial free energies in hcp metals: A molecular dynamics study of Mg. Phys. Rev. B 2006, 73, 024116. [Google Scholar] [CrossRef]
- Cahn, J.W.; Hanneman, R.E. (111) Surface tensions of III–V compounds and their relationship to spontaneous bending of thin crystals. Surf. Sci. 1964, 1, 387–398. [Google Scholar] [CrossRef]
- Wilson, S.R.; Mendelev, M.I. A unified relation for the solid-liquid interface free energy of pure FCC, BCC, and HCP metals. J. Chem. Phys. 2016, 144, 144707. [Google Scholar] [CrossRef]
- Xing, M.; Pathak, A.-D.; Sanyal, S.; Peng, Q.; Liu, X.; Wen, X. Temperature-dependent surface free energy and the Wulff shape of iron and iron carbide nanoparticles: A molecular dynamics study. Appl. Surf. Sci. 2020, 509, 144859. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Crystals | Potentials | C11 | C12 | C44 |
|---|---|---|---|---|
| Cu | Ref. [15] | 167 | 124 | 76 |
| Cu_u3.eam | 167.3 | 124.2 | 76.4 | |
| Ni | Ref. [15] | 233 | 154 | 128 |
| Ni_u3.eam | 233.3 | 154.3 | 127.6 | |
| Pd | Ref. [15] | 218 | 184 | 65 |
| Pd_u3.eam | 218.2 | 184.5 | 64.9 | |
| Ag | Ref. [15] | 129 | 91 | 57 |
| Ag_u3.eam | 129.1 | 91.0 | 56.8 | |
| Mo | Ref. [16] | 464.7 | 161.5 | 108.9 |
| mo.fs.eam.alloy | 465.9 | 161.7 | 108.9 | |
| W | Ref. [16] | 522.4 | 204.4 | 160.6 |
| W.eam.fs | 534.3 | 210.2 | 164.9 | |
| Si | Ref. [43] | 151.4 | 76.4 | 56.4 |
| Si.sw | 151.6 | 76.5 | 56.5 | |
| GaAs | Ref. [44] | 123.6 | 48.2 | 39.4 |
| GaAs.tersoff | 123.9 | 48.3 | 39.2 | |
| GaN(zincblende) | Ref. [45] | 287 | 169 | 128 |
| GaN.tersoff | 287.2 | 169.1 | 128.5 |
| Crystals | Potentials | C11 | C33 | C12 | C13 | C66 |
|---|---|---|---|---|---|---|
| Mg | Ref. [46] | 69.6 | 69.5 | 25.3 | 16 | 12.8 |
| Mg_mm.eam.fs | 69.1 | 69.4 | 26.0 | 16.1 | 12.8 | |
| Zr | Ref. [22] | 150 | 175 | 85 | 67 | 36.0 |
| Zr.eam.fs | 150.9 | 177.0 | 84.5 | 70.3 | 35.8 | |
| Ti | Ref. [21] | 180.0 | 217.1 | 87.4 | 76.6 | 51.4 |
| Ti_v2.eam.fs | 179.0 | 216.8 | 85.5 | 74.7 | 51.3 | |
| GaN(wurzite) | Ref. [45] | 347 | 381 | 154 | 123 | 81 |
| GaN.tersoff | 345.4 | 380.4 | 158.1 | 123.2 | 82.2 |
| Cu | Ni | Pd | Ag | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. [15] | Ref. [40] | Cu_u3.eam | Ref. [15] | Ref. [40] | Ni_u3.eam | Ref. [15] | Ref. [40] | Pd_u3.eam | Ref. [15] | Ref. [40] | Ag_u3.eam | |
| (100) | ||||||||||||
| 1.28 | 1.288 | 1.288 | 1.58 | 1.572 | 1.570 | 1.37 | 1.377 | 1.364 | 0.705 | 0.703 | 0.702 | |
| 1.396 | 1.396 | 1.321 | 1.319 | 1.981 | 2.000 | 0.816 | 0.815 | |||||
| −0.712 | −0.748 | −0.865 | −0.771 | −2.36 | −2.706 | −1.245 | −1.353 | |||||
| 5.914 | 5.885 | 10.722 | 10.600 | 2.611 | 2.089 | 3.343 | 3.331 | |||||
| −0.992 | −0.283 | −0.927 | −0.282 | −3.25 | −2.366 | −1.666 | −1.260 | |||||
| (110) | ||||||||||||
| 1.4 | 1.413 | 1.412 | 1.73 | 1.721 | 1.719 | 1.49 | 1.482 | 1.478 | 0.77 | 0.768 | 0.764 | |
| 1.126 | 1.125 | 1.054 | 1.062 | 1.23 | 1.337 | 0.492 | 0.503 | |||||
| 0.993 | 0.992 | 0.706 | 0.710 | 1.656 | 1.771 | 0.684 | 0.690 | |||||
| −7.798 | −10.656 | −13.031 | −17.581 | −4.775 | −7.561 | −5.51 | −7.741 | |||||
| −2.263 | −3.933 | 0.95 | −2.288 | −6.654 | −8.697 | −2.246 | −3.923 | |||||
| −3.6 | −5.694 | −5.045 | −8.872 | −2.086 | −4.461 | −2.332 | −4.226 | |||||
| −4.436 | −3.980 | −7.827 | −7.604 | −3.378 | −2.552 | −3.296 | −2.965 | |||||
| (111) | ||||||||||||
| 1.17 | 1.181 | 1.181 | 1.45 | 1.436 | 1.436 | 1.22 | 1.224 | 1.215 | 0.62 | 0.62 | 0.617 | |
| 0.866 | 0.863 | 0.457 | 0.451 | 1.848 | 1.899 | 0.636 | 0.633 | |||||
| 2.054 | 1.922 | 6.526 | 6.281 | −2.914 | −3.531 | 0.888 | 0.749 | |||||
| 1.086 | 1.133 | 3.986 | 3.857 | −1.014 | −0.915 | 1.194 | 1.116 | |||||
| −1.071 | 0.798 | −1.188 | 1.450 | −2.354 | −0.367 | 1.173 | 0.119 |
| Mo | W | Si | GaAs | GaN(zincblende) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ref. [19] | mo.fs.eam.alloy | Ref. [19] | W.eam.fs | Ref. [47] | Si.sw | Ref. [47] | Ref. [28] | GaAs.tersoff | GaN.tersoff | |
| (100) | ||||||||||
| 2.1 | 2.100 | 2.924 | 2.975 | 2.53 | 2.356 | 2.2 | 1.429 | 3.380 | ||
| 2.241 | 2.240 | 3.032 | 3.393 | 0.002 | 0.524 | −0.636 | ||||
| −18.862 | −13.794 | −11.423 | −11.465 | −13.906 | ||||||
| −16.093 | −14.505 | −1.242 | −1.416 | −0.592 | ||||||
| −2.168 | −6.993 | −3.832 | −2.153 | −7.436 | ||||||
| (110) | ||||||||||
| 1.829 | 1.829 | 2.575 | 2.608 | 1.78 | 1.666 | 1.5 | 0.91 | 0.924 | 2.307 | |
| 2.019 | 2.019 | 2.385 | 2.489 | 0.005 | 0.521 | −0.852 | ||||
| 0.775 | 0.774 | 0.271 | 0.258 | 0.003 | 0.988 | −1.527 | ||||
| −3.568 | −0.257 | −16.350 | −15.639 | −22.743 | ||||||
| −9.262 | −9.778 | −6.163 | −4.037 | −6.115 | ||||||
| 7.617 | 9.342 | −8.444 | −7.475 | −13.152 | ||||||
| −0.707 | 0.476 | −4.699 | −2.067 | −10.133 | ||||||
| (111) | ||||||||||
| 2.356 | 3.337 | 1.46 | 1.361 | 1.3 | 0.751 | 1.864 | ||||
| 0.801 | 0.392 | 0.001 | 0.5 | 0.988 | −1.502 | |||||
| −20.118 | −25.246 | −3.829 | −3.058 | 1.139 | ||||||
| 6.041 | 3.730 | −3.836 | −5.056 | −1.472 | ||||||
| −12.560 | −14.568 | −7.574 | 1.486 | 0.572 |
| Mg | Zr | Ti | GaN(wurzite) | |||||
|---|---|---|---|---|---|---|---|---|
| Ref. [48] | Mg_mm.eam.fs | Ref. [22] | Zr mm.eam.fs | Ref. [21] | Ti_v2.eam.fs | Ref. [30] | GaN.tersoff | |
| (0001) | ||||||||
| 0.238 | 0.238 | 1.022 | 1.022 | 0.993 | 0.998 | 2.0027 | 1.865 | |
| 0.289 | 1.161 | 1.140 | 0.96 | 0.959 | −1.502 | |||
| 1.041 | −0.347 | −0.191 | 0.791 | |||||
| 0.947 | 0.502 | 0.616 | −1.094 | |||||
| 0.148 | 0.231 | 0.138 | 0.247 | |||||
| 0.263 | 1.086 | 1.086 | 1.061 | 1.061 | 1.7624 | 1.995 | ||
| 0.063 | 0.655 | 0.504 | 0.481 | 0.477 | −1.348 | |||
| 0.168 | 0.476 | 0.666 | 0.68 | 0.648 | −1.354 | |||
| −0.359 | −9.039 | −2.991 | −1.003 | |||||
| −2.158 | −8.234 | −10.369 | −18.547 | |||||
| 0.791 | −5.305 | −2.913 | −4.437 | |||||
| −3.594 | 1.214 | −0.055 | −12.293 | |||||
| 0.343 | 1.23 | 1.232 | 1.187 | 1.188 | 1.9707 | 2.315 | ||
| 0.123 | 0.844 | 0.905 | 0.915 | 0.916 | −0.906 | |||
| 0.317 | 0.989 | 1.010 | 1.019 | 1.018 | −1.303 | |||
| −7.452 | −16.977 | −16.299 | −27.498 | |||||
| −3.681 | −9.752 | −12.866 | −22.276 | |||||
| −1.649 | −7.660 | −7.211 | −10.043 | |||||
| −0.194 | −2.688 | −3.642 | −5.771 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Z.; Chen, Y.; Ye, W. Calculation of Surface Properties of Cubic and Hexagonal Crystals through Molecular Statics Simulations. Crystals 2020, 10, 329. https://doi.org/10.3390/cryst10040329
Tang Z, Chen Y, Ye W. Calculation of Surface Properties of Cubic and Hexagonal Crystals through Molecular Statics Simulations. Crystals. 2020; 10(4):329. https://doi.org/10.3390/cryst10040329
Chicago/Turabian StyleTang, Zihan, Yue Chen, and Wei Ye. 2020. "Calculation of Surface Properties of Cubic and Hexagonal Crystals through Molecular Statics Simulations" Crystals 10, no. 4: 329. https://doi.org/10.3390/cryst10040329
APA StyleTang, Z., Chen, Y., & Ye, W. (2020). Calculation of Surface Properties of Cubic and Hexagonal Crystals through Molecular Statics Simulations. Crystals, 10(4), 329. https://doi.org/10.3390/cryst10040329