The Design Strategy for an Aggregation- and Crystallization-Induced Emission-Active Molecule Based on the Introduction of Skeletal Distortion by Boron Complexation with a Tridentate Ligand


Abstract
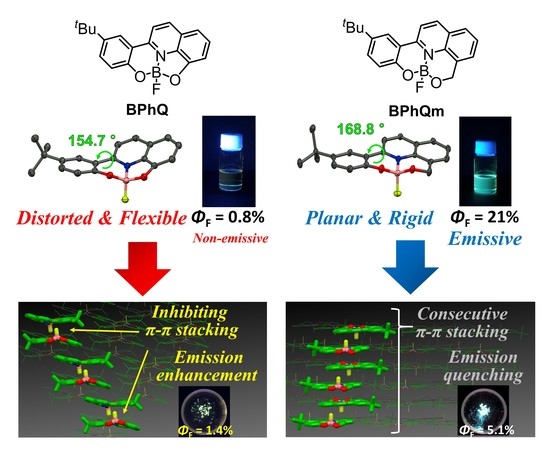
:
1. Introduction
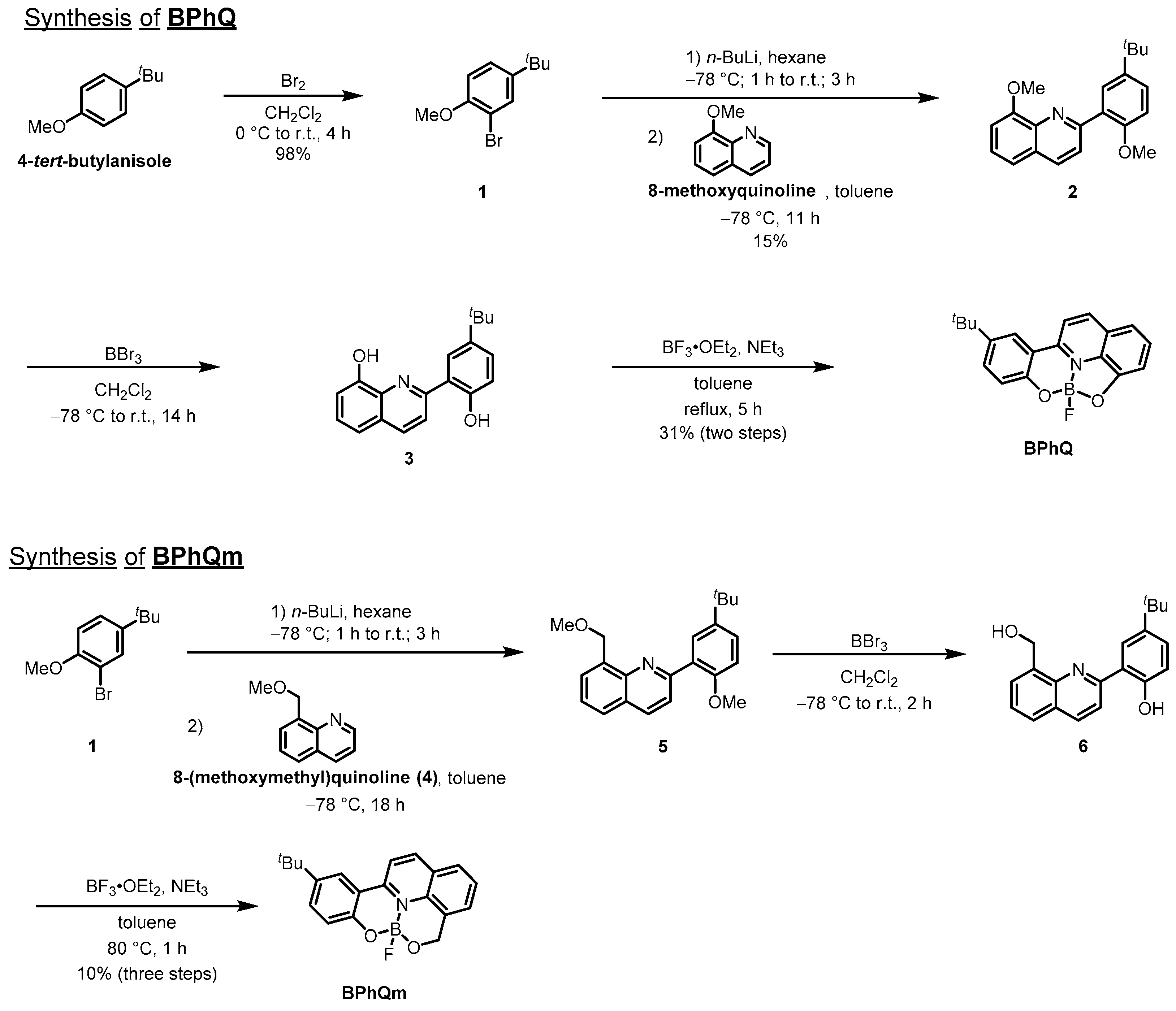
2. Materials and Methods
3. Results and Discussion
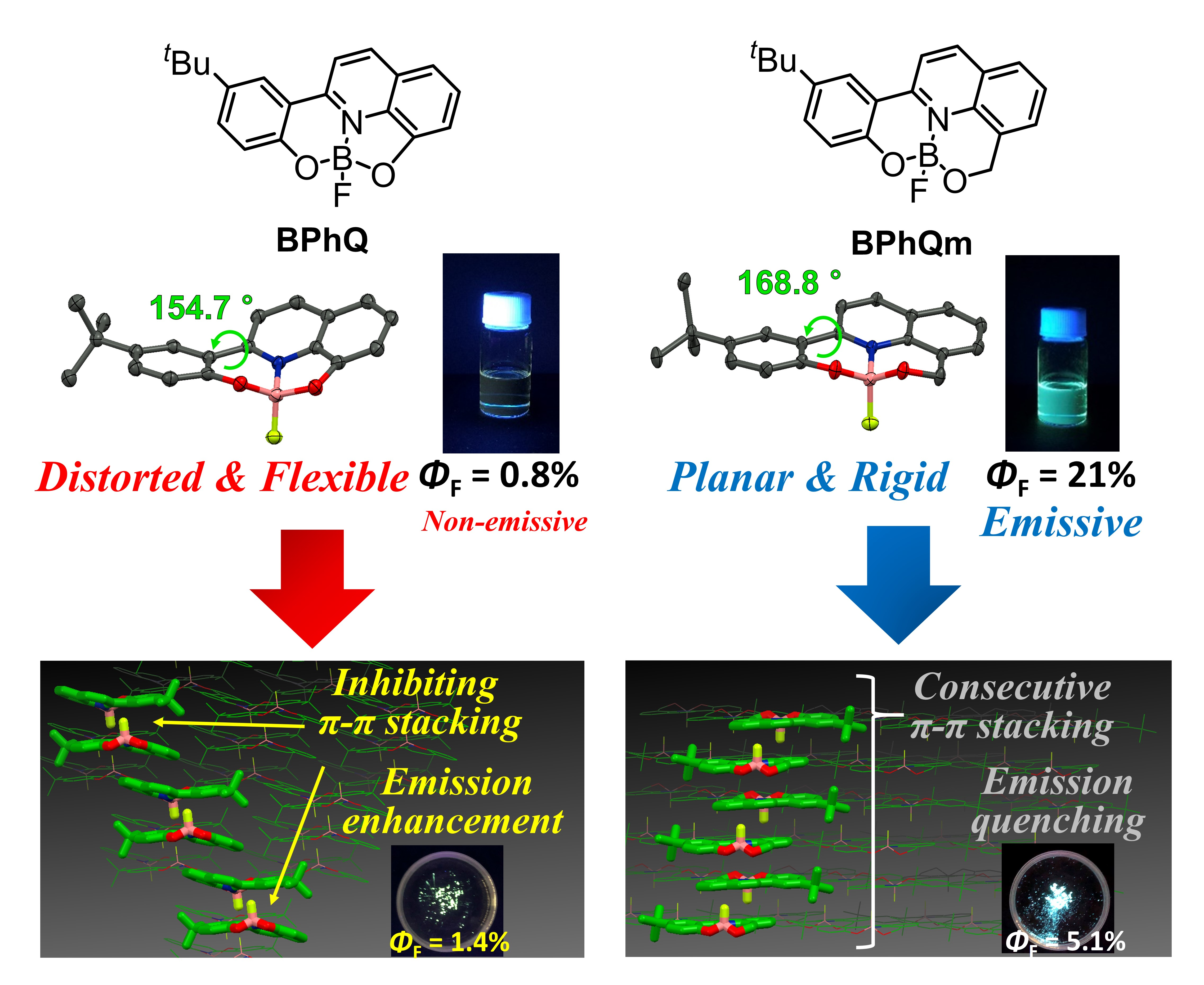
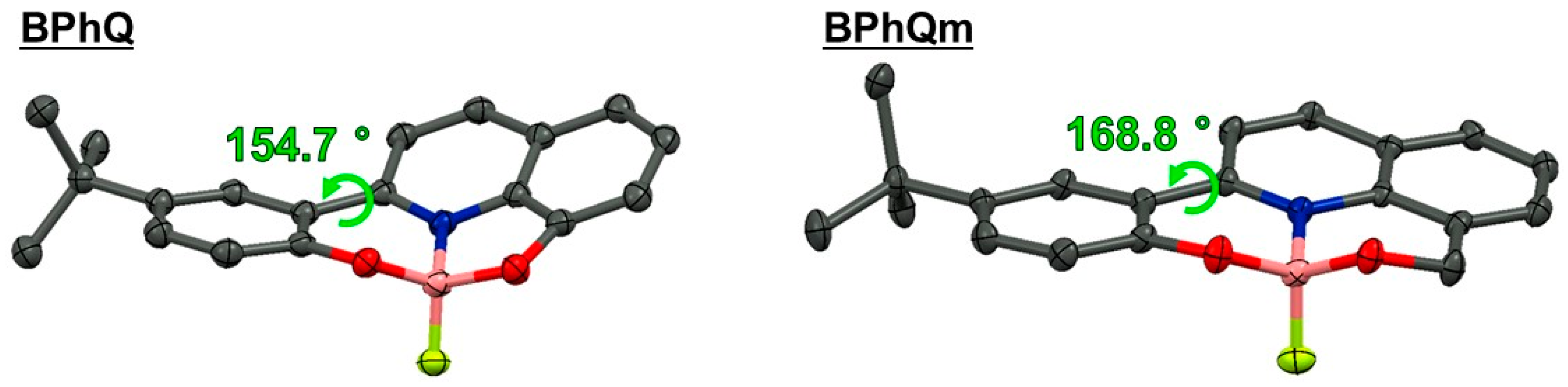
3.1. Single Crystal X-ray Diffraction Analysis
3.2. Optical Properties
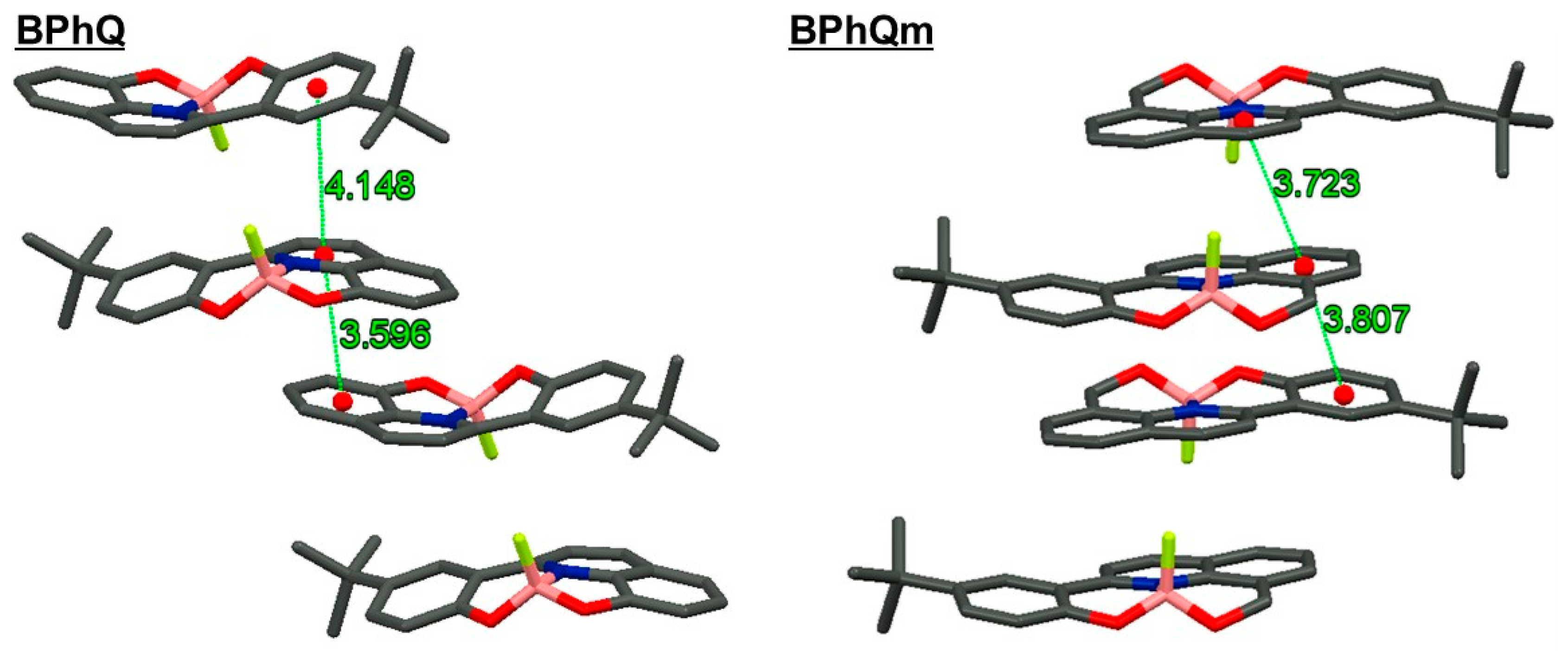
3.3. Packing Structures
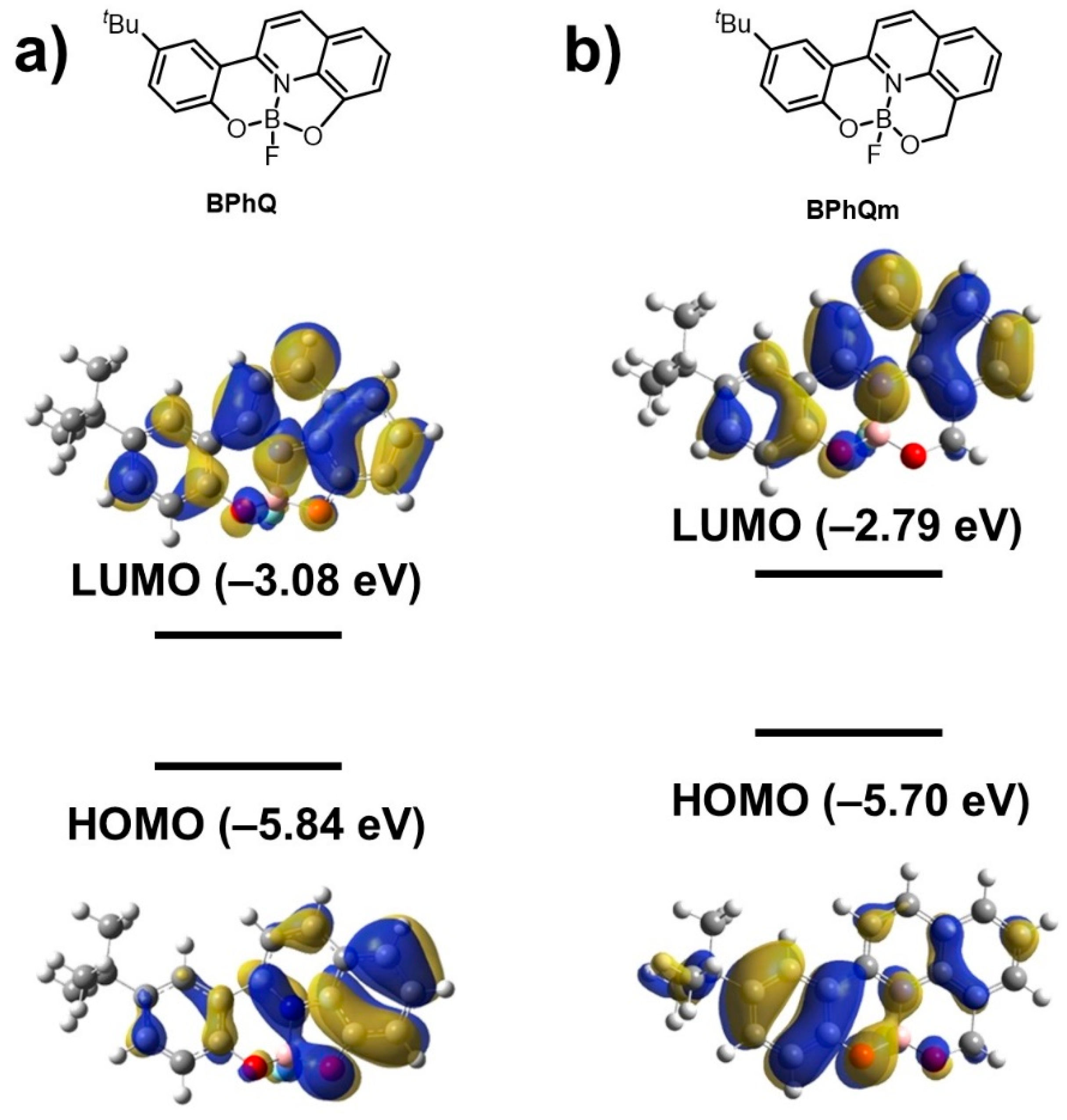
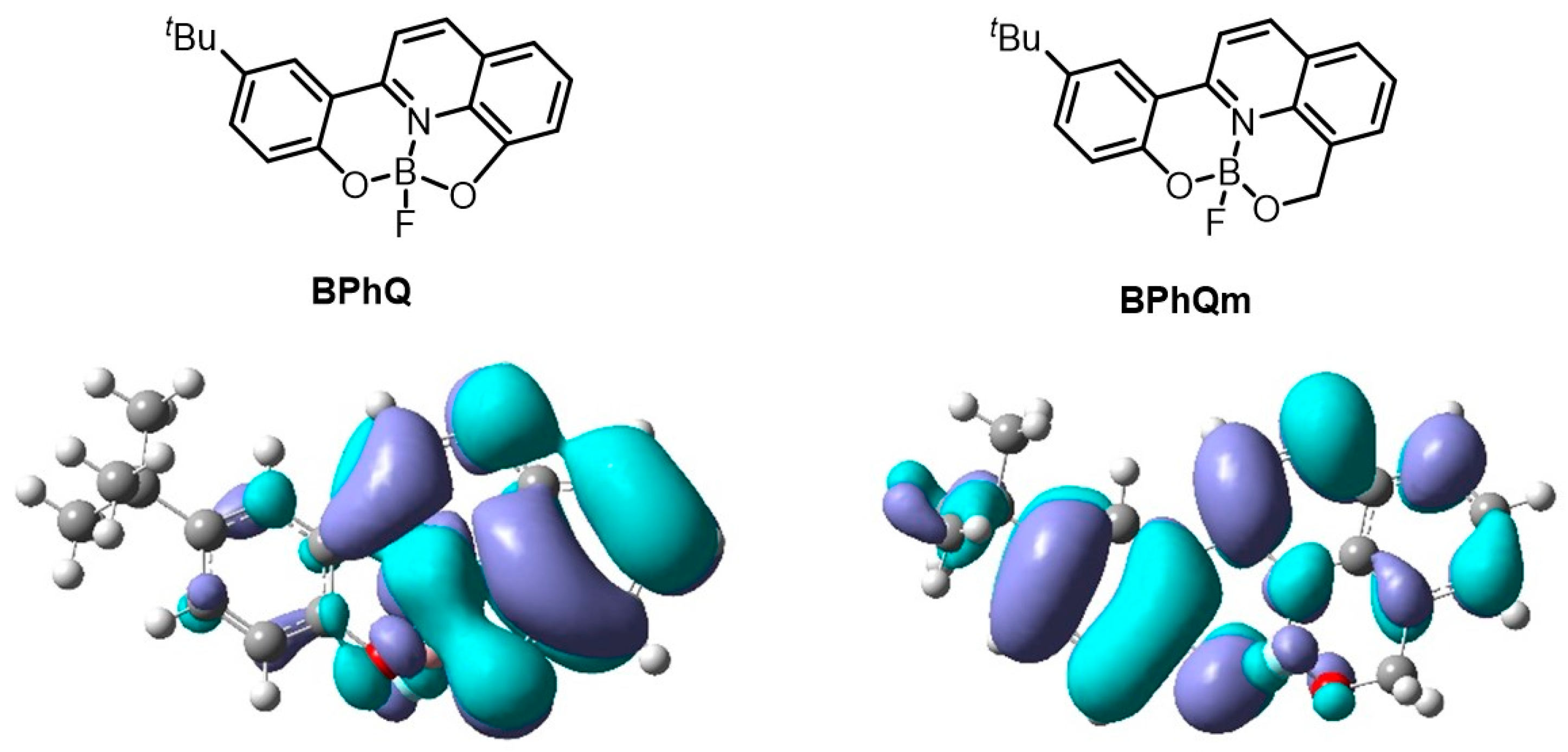
3.4. Theoretical Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yuan, L.; Lin, W.; Zheng, K.; He, L.; Huang, W. Far-red to near infrared analyte-responsive fluorescent probes based on organic fluorophore platforms for fluorescence imaging. Chem. Soc. Rev. 2013, 42, 622–661. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Tan, H.; Xie, Z.; Zhang, L.; Jing, X.; Sun, Z. Fast Response and High Sensitivity Europium Metal Organic Framework Fluorescent Probe with Chelating Terpyridine Sites for Fe3+. ACS Appl. Mater. Interfaces 2013, 5, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.S.T. Fluorescent Labeling of Biomolecules with Organic Probes. Chem. Rev. 2009, 109, 190–212. [Google Scholar] [CrossRef]
- Thompson, B.C.; Madrigal, L.G.; Pinto, M.R.; Kang, T.; Schanze, K.S.; Reynolds, J.R. Donor–acceptor copolymers for red-and near-infrared-emitting polymer light-emitting diodes. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 1417–1431. [Google Scholar] [CrossRef]
- Dias, F.B.; Bourdakos, K.N.; Jankus, V.; Moss, K.C.; Kamtekar, K.T.; Bhalla, V.; Santos, J.; Bryce, M.R.; Monkman, A.P. Triplet Harvesting with 100% Efficiency by Way of Thermally Activated Delayed Fluorescence in Charge Transfer OLED Emitters. Adv. Mater. 2013, 25, 3707–3714. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.-Y.; Ho, C.-L. Functional metallophosphors for effective charge carrier injection/transport: New robust OLED materials with emerging applications. J. Mater. Chem. 2009, 19, 4457–4482. [Google Scholar] [CrossRef]
- Ding, L.; Dong, S.-C.; Jiang, Z.-Q.; Chen, H.; Liao, L.-S. Orthogonal Molecular Structure for Better Host Material in Blue Phosphorescence and Larger OLED White Lighting Panel. Adv. Funct. Mater. 2015, 25, 645–650. [Google Scholar] [CrossRef]
- Chujo, Y.; Tanaka, K. New Polymeric Materials Based on Element-Blocks. Bull. Chem. Soc. Jpn. 2015, 88, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Gon, M.; Tanaka, K.; Chujo, Y. Recent progress in the development of advanced element-block materials. Polym. J. 2017, 50, 109–126. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Chujo, Y. Modulation of the solid-state luminescent properties of conjugated polymers by changing the connecting points of flexible boron element blocks. Polym. J. 2020, 52, 555–566. [Google Scholar] [CrossRef]
- Matsumura, Y.; Ishidoshiro, M.; Irie, Y.; Imoto, H.; Naka, K.; Tanaka, K.; Inagi, S.; Tomita, I. Arsole-Containing π-Conjugated Polymer by the Post-Element-Transformation Technique. Angew. Chem. Int. Ed. 2016, 55, 15040–15043. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, J.; Gon, M.; Tanaka, K.; Chujo, Y. A Near-Infrared Absorptive and Emissive Poly(p-phenylene vinylene) Derivative Containing Azobenzene–Boron Complexes. Macromolecules 2020, 53, 4524–4532. [Google Scholar] [CrossRef]
- Ohtani, S.; Nakamura, M.; Gon, M.; Tanaka, K.; Chujo, Y. Synthesis of Fully-Fused Bisboron Azomethine Complexes and Their Conjugated Polymers with Solid-State Near-Infrared Emission. Chem. Commun. 2020, 56, 6575–6578. [Google Scholar] [CrossRef] [PubMed]
- Gon, M.; Wakabayashi, J.; Tanaka, K.; Chujo, Y. Unique Substitution Effect at 5,5′-Positions of Fused Azobenzene–Boron Complexes with a N=N π-Conjugated System. Chem. Asian J. 2019, 14, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, S.; Gon, M.; Tanaka, K.; Chujo, Y. Construction of the Luminescent Donor–Acceptor Conjugated Systems Based on Boron-Fused Azomethine Acceptor. Macromolecules 2019, 52, 3387–3393. [Google Scholar] [CrossRef]
- Gon, M.; Tanaka, K.; Chujo, Y. A Highly Efficient Near-Infrared-Emissive Copolymer with a N=N Double-Bond π-Conjugated System Based on a Fused Azobenzene-Boron Complex. Angew. Chem. Int. Ed. 2018, 57, 6546–6551. [Google Scholar] [CrossRef]
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef]
- Ulrich, G.; Ziessel, R.; Harriman, A. The Chemistry of Fluorescent Bodipy Dyes: Versatility Unsurpassed. Angew. Chem. Int. Ed. 2008, 47, 1184–1201. [Google Scholar] [CrossRef]
- Frath, D.; Azizi, S.; Ulrich, G.; Retailleau, P.; Ziessel, R. Facile Synthesis of Highly Fluorescent Boranil Complexes. Org. Lett. 2011, 13, 3414–3417. [Google Scholar] [CrossRef]
- Frath, D.; Massue, J.; Ulrich, G.; Ziessel, R. Luminescent Materials: Locking π-Conjugated and Heterocyclic Ligands with Boron(III). Angew. Chem. Int. Ed. 2014, 53, 2290–2310. [Google Scholar] [CrossRef]
- Suresh, D.; Ferreira, B.; Lopes, P.S.; Gomes, C.S.B.; Krishnamoorthy, P.; Charas, A.; Vila-Viçosa, D.; Morgado, J.; Calhorda, M.J.; Maçanita, A.L.; et al. Boron complexes of aromatic ring fused iminopyrrolyl ligands: Synthesis, structure, and luminescence properties. Dalton Trans. 2016, 45, 15603–15620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chen, J.; Payne, S.J.; Kooi, S.E.; Demas, J.N.; Fraser, C.L. Multi-Emissive Difluoroboron Dibenzoylmethane Polylactide Exhibiting Intense Fluorescence and Oxygen-Sensitive Room-Temperature Phosphorescence. J. Am. Chem. Soc. 2007, 129, 8942–8943. [Google Scholar] [CrossRef] [PubMed]
- Hesari, M.; Barbon, S.M.; Staroverov, V.N.; Ding, Z.; Gilroy, J.B. Efficient electrochemiluminescence of a readily accessible boron difluoride formazanate dye. Chem. Commun. 2015, 51, 3766–3769. [Google Scholar] [CrossRef] [PubMed]
- Vuong, T.M.H.; Weimmerskirch-Aubatin, J.; Lohier, J.-F.; Bar, N.; Boudin, S.; Labbé, C.; Gourbilleau, F.; Nguyen, H.; Dang, T.T.; Villemin, D. Blue highly fluorescent boron difluoride complexes based on phthalazine–pyridine. New J. Chem. 2016, 40, 6070–6076. [Google Scholar] [CrossRef]
- Ando, N.; Soutome, H.; Yamaguchi, S. Near-infrared fluorescein dyes containing a tricoordinate boron atom. Chem. Sci. 2019, 10, 7816–7821. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, K.; Huang, S.; Qu, S.; Liu, X.; Zhu, Q.; Zhang, H.; Wang, Y. Brightly fluorescent red organic solids bearing boron-bridged π–conjugated skeletons. J. Mater. Chem. 2011, 21, 15298–15304. [Google Scholar] [CrossRef]
- Urban, M.; Durka, K.; Górka, P.; Wiosna-Sałyga, G.; Nawara, K.; Jankowski, P.; Luliński, S. The effect of locking π-conjugation in organoboron moieties in the structures of luminescent tetracoordinate boron complexes. Dalton Trans. 2019, 48, 8642–8663. [Google Scholar] [CrossRef]
- Wakamiya, A.; Taniguchi, T.; Yamaguchi, S. Intramolecular B–N Coordination as a Scaffold for Electron-Transporting Materials: Synthesis and Properties of Boryl-Substituted Thienylthiazoles. Angew. Chem. Int. Ed. 2006, 45, 3170–3173. [Google Scholar] [CrossRef]
- Yoshino, J.; Kano, N.; Kawashima, T. Synthesis of the most intensely fluorescent azobenzene by utilizing the B–N interaction. Chem. Commun. 2007, 559–561. [Google Scholar] [CrossRef]
- Yoshino, J.; Kano, N.; Kawashima, T. Fluorescence Properties of Simple N-Substituted Aldimines with a B−N Interaction and Their Fluorescence Quenching by a Cyanide Ion. J. Org. Chem. 2009, 74, 7496–7503. [Google Scholar] [CrossRef]
- Kwok, R.T.K.; Leung, C.W.T.; Lam, J.W.Y.; Tang, B.Z. Biosensing by luminogens with aggregation-induced emission characteristics. Chem. Soc. Rev. 2015, 44, 4228–4238. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Leung, N.L.C.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xie, Z.; Lam, J.W.Y.; Cheng, L.; Chen, H.; Qiu, C.; Kwok, H.S.; Zhan, X.; Liu, Y.; Zhu, D.; et al. Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chem. Commun. 2001, 1740–1741. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Lam, J.W.Y.; Tang, B.Z. Aggregation-induced emission. Chem. Soc. Rev. 2011, 40, 5361–5388. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Lam, J.W.Y.; Qin, A.; Li, Z.; Sun, J.; Sung, H.H.-Y.; Williams, I.D.; Tang, B.Z. Switching the light emission of (4-biphenylyl)phenyldibenzofulvene by morphological modulation: Crystallization-induced emission enhancement. Chem. Commun. 2007, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Galer, P.; Korošec, R.C.; Vidmar, M.; Šket, B. Crystal Structures and Emission Properties of the BF2 Complex 1-Phenyl-3-(3,5-dimethoxyphenyl)-propane-1,3-dione: Multiple Chromisms, Aggregation- or Crystallization-Induced Emission, and the Self-Assembly Effect. J. Am. Chem. Soc. 2014, 136, 7383–7394. [Google Scholar] [CrossRef]
- Yoshii, R.; Hirose, A.; Tanaka, K.; Chujo, Y. Functionalization of Boron Diiminates with Unique Optical Properties: Multicolor Tuning of Crystallization-Induced Emission and Introduction into the Main Chain of Conjugated Polymers. J. Am. Chem. Soc. 2014, 136, 18131–18139. [Google Scholar] [CrossRef]
- Gon, M.; Tanaka, K.; Chujo, Y. Concept of Excitation-Driven Boron Complexes and Their Applications for Functional Luminescent Materials. Bull. Chem. Soc. Jpn. 2019, 92, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Nishino, K.; Ito, S.; Yamane, H.; Suenaga, K.; Hashimoto, K.; Chujo, Y. Development of Solid-State Emissive o-Carborane and Theoretical Investigation of the Mechanism of the Aggregation-Induced Emission Behaviors of Organoboron “Element-Blocks”. Faraday Discuss. 2017, 196, 31–42. [Google Scholar] [CrossRef]
- Ohtani, S.; Gon, M.; Tanaka, K.; Chujo, Y. A Flexible, Fused, Azomethine–Boron Complex: Thermochromic Luminescence and Thermosalient Behavior in Structural Transitions between Crystalline Polymorphs. Chem. Eur. J. 2017, 23, 11827–11833. [Google Scholar] [CrossRef]
- Ma, R.-Z.; Yao, Q.-C.; Yang, X.; Xia, M. Synthesis, characterization and photoluminescence properties of strong fluorescent BF2 complexes bearing (2-quinolin-2-yl) phenol ligands. J. Fluorine Chem. 2012, 137, 93–98. [Google Scholar] [CrossRef]
- Tokoro, Y.; Nagai, A.; Kokado, K.; Chujo, Y. Synthesis of Organoboron Quinoline-8-thiolate and Quinoline-8-selenolate Complexes and Their Incorporation into the π-Conjugated Polymer Main-Chain. Macromolecules 2009, 42, 2988–2993. [Google Scholar] [CrossRef]
- Balijapalli, U.; Iyer, S.K. Synthesis and Optical Properties of a Series of Green-Light-Emitting 2-(4-Phenylquinolin-2-yl)phenol–BF2 Complexes (Boroquinols). Eur. J. Org. Chem. 2015, 2015, 5089–5098. [Google Scholar] [CrossRef]
- Qi, Y.; Kang, R.; Huang, J.; Zhang, W.; He, G.; Yin, S.; Fang, Y. Reunderstanding the Fluorescent Behavior of Four-Coordinate Monoboron Complexes Containing Monoanionic Bidentate Ligands. J. Phys. Chem. B 2017, 121, 6189–6199. [Google Scholar] [CrossRef]
- Shi, J.; Aguilar Suarez, L.E.; Yoon, S.-J.; Varghese, S.; Serpa, C.; Park, S.Y.; Lüer, L.; Roca-Sanjuán, D.; Milián-Medina, B.; Gierschner, J. Solid State Luminescence Enhancement in π-Conjugated Materials: Unraveling the Mechanism beyond the Framework of AIE/AIEE. J. Phys. Chem. C 2017, 121, 23166–23183. [Google Scholar] [CrossRef]
- Ma, L.; Yu, Y.; Jiao, B.; Hou, X.; Wu, Z. Theoretical evidence of low-threshold amplified spontaneous emission in organic emitters: Transition density and intramolecular vibrational mode analysis. Phys. Chem. Chem. Phys. 2018, 20, 19515–19524. [Google Scholar] [CrossRef]
- Shizu, K.; Noda, H.; Tanaka, H.; Taneda, M.; Uejima, M.; Sato, T.; Tanaka, K.; Kaji, H.; Adachi, C. Highly Efficient Blue Electroluminescence Using Delayed-Fluorescence Emitters with Large Overlap Density between Luminescent and Ground States. J. Phys. Chem. C 2015, 119, 26283–26289. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λmax,abs (nm) a | ε (105M–1cm–1) b | λem (nm) c | ΦFd | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sol. a | Agg. e | Film f | Cryst. g | Sol. a | Agg. e | Film f | Cryst. g | |||
| BPhQ | 377 | 0.11 | 550 | 537 | 531 | 518 | 0.008 | 0.014 | 0.021 | 0.017 |
| BPhQm | 387 | 0.097 | 493 | 460 | 487 | 479 | 0.21 | 0.072 | 0.16 | 0.051 |
| Compound | τ (ns) a | kr (×108s−1) b | knr (×108s−1) b | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sol. c | Agg. d | Film e | Cryst. f | Sol. c | Agg. d | Film e | Cryst. f | Sol. c | Agg. d | Film e | Cryst. f | |
| BPhQ | 0.6 | 0.3 (4%) 1.7 (38%) 4.3 (58%) | 2.1 (32%) 5.8 (68%) | 2.0 (66%) 3.3 (34%) | 0.12 | 0.038 | 0.040 | 0.065 | 15 | 2.7 | 1.9 | 3.8 |
| BPhQm | 2.8 | 1.2 (71%) 3.9 (29%) | 0.6 (8%) 1.7 (70%) 3.8 (23%) | 0.4 (24%) 1.0 (64%) 3.0 (12%) | 0.74 | 0.26 | 0.64 | 0.32 | 2.8 | 3.3 | 3.3 | 5.9 |
| Compound | Excitation Energy | S1→S0 Transition Contribution | Oscillator Strength | μ a |
|---|---|---|---|---|
| BPhQ | 2.30 eV (540 nm) | HOMO–LUMO (94%) | 0.0262 | 0.4656 a.u. |
| BPhQm | 2.57 eV (482 nm) | HOMO–LUMO (95%) | 0.1521 | 2.415 a.u. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohtani, S.; Gon, M.; Tanaka, K.; Chujo, Y. The Design Strategy for an Aggregation- and Crystallization-Induced Emission-Active Molecule Based on the Introduction of Skeletal Distortion by Boron Complexation with a Tridentate Ligand. Crystals 2020, 10, 615. https://doi.org/10.3390/cryst10070615
Ohtani S, Gon M, Tanaka K, Chujo Y. The Design Strategy for an Aggregation- and Crystallization-Induced Emission-Active Molecule Based on the Introduction of Skeletal Distortion by Boron Complexation with a Tridentate Ligand. Crystals. 2020; 10(7):615. https://doi.org/10.3390/cryst10070615
Chicago/Turabian StyleOhtani, Shunsuke, Masayuki Gon, Kazuo Tanaka, and Yoshiki Chujo. 2020. "The Design Strategy for an Aggregation- and Crystallization-Induced Emission-Active Molecule Based on the Introduction of Skeletal Distortion by Boron Complexation with a Tridentate Ligand" Crystals 10, no. 7: 615. https://doi.org/10.3390/cryst10070615
APA StyleOhtani, S., Gon, M., Tanaka, K., & Chujo, Y. (2020). The Design Strategy for an Aggregation- and Crystallization-Induced Emission-Active Molecule Based on the Introduction of Skeletal Distortion by Boron Complexation with a Tridentate Ligand. Crystals, 10(7), 615. https://doi.org/10.3390/cryst10070615