Making NSCLC Crystal Clear: How Kinase Structures Revolutionized Lung Cancer Treatment




Abstract
:
1. Introduction
2. Kinases: A Structural Overview
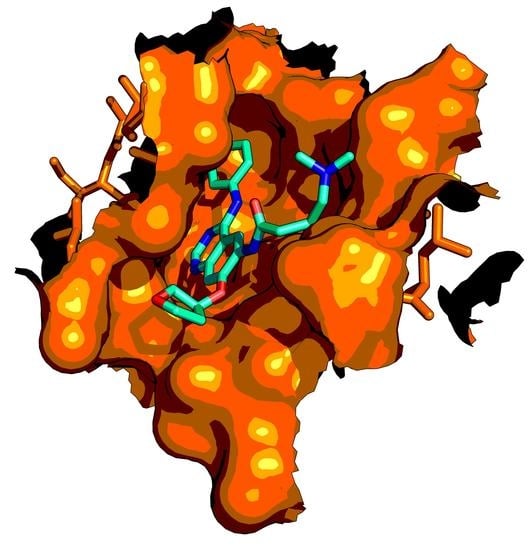
3. Epidermal Growth Factor Receptor (EGFR)
4. Anaplastic Lymphoma Kinase (ALK)
5. Rapidly Accelerated Fibrosarcoma Homologue B (BRAF)
6. Kirsten Rat Sarcoma Viral Oncogene Homologue (KRAS)
7. Molecular Modeling as a Supporting Tool for Personalized Medicine: The Future Is Knocking on the Door
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.D. Lung Cancer Pathology. Clin. Chest Med. 2020, 41, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-L.; Yuan, J.-Q.; Wang, K.-F.; Fu, X.-H.; Han, X.-R.; Threapleton, D.; Yang, Z.-Y.; Mao, C.; Tang, J.-L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [Green Version]
- Brustugun, O.T.; Khattak, A.M.; Trømborg, A.K.; Beigi, M.; Beiske, K.; Lund-Iversen, M.; Helland, Å. BRAF-mutations in non-small cell lung cancer. Lung Cancer 2014, 84, 36–38. [Google Scholar] [CrossRef]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the “undruggable”. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118570. [Google Scholar] [CrossRef]
- Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemother. Res. Pract. 2014, 2014. [Google Scholar] [CrossRef]
- Pimentel, A.S.; Guimarães, C.R.W.; Miller, Y. Molecular Modeling: Advancements and Applications. J. Chem. 2013, 2013, 1–2. [Google Scholar] [CrossRef]
- Smondyrev, A.M.; Voth, G.A. Molecular dynamics simulation of proton transport tnrougn tne influenza A virus M2 channel. Biophys. J. 2002, 83, 1987–1996. [Google Scholar] [CrossRef] [Green Version]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z. Advances in Homology Protein Structure Modeling. Curr. Protein Pept. Sci. 2006, 7, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. Des. Sel. 1999, 12, 85–94. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Camacho, C.J. PocketQuery: Protein-protein interaction inhibitor starting points from protein-protein interaction structure. Nucleic Acids Res. 2012, 40, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Dömling, A.; Camacho, C.J. AnchorQuery: Rapid online virtual screening for small-molecule protein-protein interaction inhibitors. Protein Sci. 2018, 27, 229–232. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [Green Version]
- Geng, C.; Narasimhan, S.; Rodrigues, J.P.G.L.M.; Bonvin, A.M.J.J. Information-driven, ensemble flexible peptide docking using HADDOCK. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1561, pp. 109–138. [Google Scholar]
- Ricci, C.G.; Netz, P.A. Docking Studies on DNA-Ligand Interactions: Building and Application of a Protocol To Identify the Binding Mode. J. Chem. Inf. Model. 2009, 49, 1925–1935. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Nerenberg, P.S.; Head-Gordon, T. New developments in force fields for biomolecular simulations. Curr. Opin. Struct. Biol. 2018, 49, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defelipe, L.A.; Arcon, J.P.; Modenutti, C.P.; Marti, M.A.; Turjanski, A.G.; Barril, X. Solvents to fragments to drugs: MD applications in drug design. Molecules 2018, 23, 3269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008, 27, 253–261. [Google Scholar] [CrossRef]
- Madhusudan; Trafny, E.A.; Xuong, N.-H.; Adams, J.A.; Eyck, L.F.T.; Taylor, S.S.; Sowadski, J.M. cAMP-dependent protein kinase: Crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 2008, 3, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Huse, M.; Kuriyan, J. The Conformational Plasticity of Protein Kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef] [Green Version]
- D’Abramo, M.; Besker, N.; Chillemi, G.; Grottesi, A. Modeling conformational transitions in kinases by molecular dynamics simulations: Achievements, difficulties, and open challenges. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Möbitz, H. The ABC of protein kinase conformations. Biochim. Biophys. Acta-Proteins Proteom. 2015, 1854, 1555–1566. [Google Scholar] [CrossRef]
- Meharena, H.S.; Chang, P.; Keshwani, M.M.; Oruganty, K.; Nene, A.K.; Kannan, N.; Taylor, S.S.; Kornev, A.P. Deciphering the Structural Basis of Eukaryotic Protein Kinase Regulation. PLoS Biol. 2013, 11, e1001680. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Doshi, A.; Lei, M.; Eck, M.J.; Harrison, S.C. Crystal Structures of c-Src Reveal Features of Its Autoinhibitory Mechanism. Mol. Cell 1999, 3, 629–638. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Kamata, N.; Takechi, M.; Mitani, Y.; Ohta, K.; Fujimoto, S.; Hiraoka, M.; Higashikawa, K.; Shigeishi, H. Expression of epiregulin, a novel epidermal growth factor ligand associated with prognosis in human oral squamous cell carcinomas. Oncol. Rep. 2008, 19, 1557–1564. [Google Scholar] [CrossRef]
- Lemmon, M.A. Ligand-induced ErbB receptor dimerization. Exp. Cell Res. 2009, 315, 638–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gureasko, J.; Galush, W.J.; Boykevisch, S.; Sondermann, H.; Bar-Sagi, D.; Groves, J.T.; Kuriyan, J. Membrane-dependent signal integration by the Ras activator Son of sevenless. Nat. Struct. Mol. Biol. 2008, 15, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.; Gee, J.M.; Harper, M. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Lee, B.; Lee, T.; Lee, S.-H.; Choi, Y.-L.; Han, J. Clinicopathologic characteristics of EGFR, KRAS, and ALK alterations in 6,595 lung cancers. Oncotarget 2016, 7, 23874–23884. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.-S.; Leahy, D.J.; Lemmon, M.A. EGF Activates Its Receptor by Removing Interactions that Autoinhibit Ectodomain Dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Dawson, J.P.; Berger, M.B.; Lin, C.-C.; Schlessinger, J.; Lemmon, M.A.; Ferguson, K.M. Epidermal Growth Factor Receptor Dimerization and Activation Require Ligand-Induced Conformational Changes in the Dimer Interface. Mol. Cell. Biol. 2005, 25, 7734–7742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purba, E.; Saita, E.; Maruyama, I. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.-H.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Jura, N.; Zhang, X.; Endres, N.F.; Seeliger, M.A.; Schindler, T.; Kuriyan, J. Catalytic Control in the EGF Receptor and Its Connection to General Kinase Regulatory Mechanisms. Mol. Cell 2011, 42, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Arkhipov, A.; Kim, E.T.; Pan, A.C.; Shaw, D.E. Transitions to catalytically inactive conformations in EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 7270–7275. [Google Scholar] [CrossRef] [Green Version]
- George Priya Doss, C.; Rajith, B.; Chakraborty, C.; NagaSundaram, N.; Ali, S.K.; Zhu, H. Structural signature of the G719S-T790M double mutation in the EGFR kinase domain and its response to inhibitors. Sci. Rep. 2014, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Saldaña-Rivera, L.; Bello, M.; Méndez-Luna, D. Structural insight into the binding mechanism of ATP to EGFR and L858R, and T790M and L858R/T790 mutants. J. Biomol. Struct. Dyn. 2019, 37, 4671–4684. [Google Scholar] [CrossRef]
- Keyloun, K.R.; Reid, M.C.; Choi, R.; Song, Y.; Fox, A.M.W.; Hillesland, H.K.; Zhang, Z.; Vidadala, R.; Merritt, E.A.; Lau, A.O.T.; et al. The gatekeeper residue and beyond: Homologous calcium-dependent protein kinases as drug development targets for veterinarian Apicomplexa parasites. Parasitology 2014, 141, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR Family: Not So Prototypical Receptor Tyrosine Kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, N.; Tojo, A.; Hino, M.; Yazaki, Y.; Shibuya, M. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 1992, 186, 768–774. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib). Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.H.; Dolder, C.R. Lapatinib: A Novel Dual Tyrosine Kinase Inhibitor with Activity in Solid Tumors. Ann. Pharmacother. 2006, 40, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Discovery Studio Visualization; Dassault Systemes BIOVIA: San Diego, CA, USA, 2016.
- Songtawee, N.; Bevan, D.R.; Choowongkomon, K. Molecular dynamics of the asymmetric dimers of EGFR: Simulations on the active and inactive conformations of the kinase domain. J. Mol. Graph. Model. 2015, 58, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016, 107, 1179–1186. [Google Scholar] [CrossRef]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28, S24–S31. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Yang, M.; Liang, N.; Li, S. Determining EGFR-TKI sensitivity of G719X and other uncommon EGFR mutations in non-small cell lung cancer: Perplexity and solution. Oncol. Rep. 2017, 37, 1347–1358. [Google Scholar] [CrossRef] [Green Version]
- Noronha, V.; Choughule, A.; Patil, V.M.; Joshi, A.; Kumar, R.; Philip, D.S.J.; Banavali, S.; Dutt, A.; Prabhash, K. Epidermal growth factor receptor exon 20 mutation in lung cancer: Types, incidence, clinical features and impact on treatment. Onco. Targets. Ther. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Avizienyte, E.; Ward, R.A.; Garner, A.P. Comparison of the EGFR resistance mutation profiles generated by EGFR-targeted tyrosine kinase inhibitors and the impact of drug combinations. Biochem. J. 2008, 415, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non–Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, S.; Kukimoto-Niino, M.; Parker, L.; Handa, N.; Terada, T.; Fujimoto, T.; Terazawa, Y.; Wakiyama, M.; Sato, M.; Sano, S.; et al. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene 2013, 32, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and Clinical Relevance of the Epidermal Growth Factor Receptor in Human Cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eck, M.J.; Yun, C.H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta-Proteins Proteomics 2010, 1804, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassunke, J.; Müller, F.; Keul, M.; Michels, S.; Dammert, M.A.; Schmitt, A.; Plenker, D.; Lategahn, J.; Heydt, C.; Brägelmann, J.; et al. Overcoming EGFRG724S-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat. Commun. 2018, 9, 4655. [Google Scholar] [CrossRef]
- Takeda, M.; Nakagawa, K. First- and Second-Generation EGFR-TKIs Are All Replaced to Osimertinib in Chemo-Naive EGFR Mutation-Positive Non-Small Cell Lung Cancer? Int. J. Mol. Sci. 2019, 20, 146. [Google Scholar] [CrossRef] [Green Version]
- El Meskini, R.; Iacovelli, A.; Kulaga, A.; Gumprecht, M.; Spencer, M.; Ileva, L.; Simmons, A.; Weaver Ohler, Z. Abstract A162: Continuous treatment with rociletinib in EGFR transgenic mice results in acquired resistance to both rociletinib and osimertinib and intra-“patient” tumor heterogeneity. In EGFR/Her2; American Association for Cancer Research: Philadelphia, PA, USA, 2018; p. A162. [Google Scholar]
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 2017, 8, 49671–49679. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.-H.I.; Cui, J.; Schrock, A.B.; Goldberg, M.E.; Zhu, V.W.; Albacker, L.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR. Lung Cancer 2017, 108, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Rangachari, D.; To, C.; Shpilsky, J.E.; VanderLaan, P.A.; Kobayashi, S.S.; Mushajiang, M.; Lau, C.J.; Paweletz, C.P.; Oxnard, G.R.; Jänne, P.A.; et al. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J. Thorac. Oncol. 2019, 14, 1995–2002. [Google Scholar] [CrossRef]
- Niederst, M.J.J.; Hu, H.; Mulvey, H.E.E.; Lockerman, E.L.L.; Garcia, A.R.R.; Piotrowska, Z.; Sequist, L.V.V.; Engelman, J.A.A. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, K.D.; Garton, A.J.; Romero, M.S.; Kahler, J.; Thomson, S.; Ross, S.; Park, F.; Haley, J.D.; Gibson, N.; Sliwkowski, M.X. Kinetic Analysis of Epidermal Growth Factor Receptor Somatic Mutant Proteins Shows Increased Sensitivity to the Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, Erlotinib. Cancer Res. 2006, 66, 8163–8171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, N.; Azuma, K.; Sakai, K.; Hattori, S.; Kawahara, A.; Ishii, H.; Tokito, T.; Kinoshita, T.; Yamada, K.; Nishio, K.; et al. Association of EGFR Exon 19 Deletion and EGFR-TKI Treatment Duration with Frequency of T790M Mutation in EGFR-Mutant Lung Cancer Patients. Sci. Rep. 2016, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bulbul, A.; Husain, H. First-Line Treatment in EGFR Mutant Non-Small Cell Lung Cancer: Is There a Best Option? Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Ou, Q.; Wu, X.; Wang, X.; Shao, Y.W.; Ying, J. Acquired EGFR L718V mutation mediates resistance to osimertinib in non-small cell lung cancer but retains sensitivity to afatinib. Lung Cancer 2018, 118, 1–5. [Google Scholar] [CrossRef]
- Liu, J.; Jin, B.; Su, H.; Qu, X.; Liu, Y. Afatinib helped overcome subsequent resistance to osimertinib in a patient with NSCLC having leptomeningeal metastasis baring acquired EGFR L718Q mutation: A case report. BMC Cancer 2019, 19, 702. [Google Scholar] [CrossRef]
- Brown, B.P.; Zhang, Y.-K.; Westover, D.; Yan, Y.; Qiao, H.; Huang, V.; Du, Z.; Smith, J.A.; Ross, J.S.; Miller, V.A.; et al. On-target resistance to the mutant-selective EGFR inhibitor osimertinib can develop in an allele specific manner dependent on the original EGFR activating mutation. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Fujino, T.; Nishino, M.; Koga, T.; Chiba, M.; Sesumi, Y.; Ohara, S.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. EGFR T790M and C797S Mutations as Mechanisms of Acquired Resistance to Dacomitinib. J. Thorac. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokudome, N.; Koh, Y.; Akamatsu, H.; Fujimoto, D.; Okamoto, I.; Nakagawa, K.; Hida, T.; Imamura, F.; Morita, S.; Yamamoto, N. Differential significance of molecular subtypes which were classified into EGFR exon 19 deletion on the first line afatinib monotherapy. BMC Cancer 2020, 20, 103. [Google Scholar] [CrossRef] [PubMed]
- Tamirat, M.Z.; Koivu, M.; Elenius, K.; Johnson, M.S. Structural characterization of EGFR exon 19 deletion mutation using molecular dynamics simulation. PLoS ONE 2019, 14, e0222814. [Google Scholar] [CrossRef] [PubMed]
- van Kempen, L.C.; Wang, H.; Aguirre, M.L.; Spatz, A.; Kasymjanova, G.; Vilacha, J.F.; Groves, M.R.; Agulnik, J.; Small, D. Afatinib in Osimertinib-Resistant EGFR ex19del/T790M/P794L Mutated NSCLC. J. Thorac. Oncol. 2018, 13, e161–e163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, D.B. Kinase inhibitor-responsive genotypes in EGFR mutated lung adenocarcinomas: Moving past common point mutations or indels into uncommon kinase domain duplications and rearrangements. Transl. Lung Cancer Res. 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Liu, X.; Song, X.; Yao, J. Chemotherapy Drug Response to the L858R-induced Conformational Change of EGFR Activation Loop in Lung Cancer. Mol. Inform. 2016, 35, 529–537. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Cheng, G.; Song, Z.; Chen, D. Clinical efficacy of first-generation EGFR-TKIs in patients with advanced non-small-cell lung cancer harboring EGFR exon 20 mutations. Oncol. Targets. Ther. 2016, 9, 4181–4186. [Google Scholar] [CrossRef] [Green Version]
- Barker, A.J.; Gibson, K.H.; Grundy, W.; Godfrey, A.A.; Barlow, J.J.; Healy, M.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Scarlett, L.; et al. Studies leading to the identification of ZD1839 (iressaTM): An orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg. Med. Chem. Lett. 2001, 11, 1911–1914. [Google Scholar] [CrossRef]
- Yasuda, H.; Park, E.; Yun, C.-H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.-L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, Biochemical, and Clinical Characterization of Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutations in Lung Cancer. Sci. Transl. Med. 2014, 5, 225er1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello, M. Binding mechanism of kinase inhibitors to EGFR and T790M, L858R and L858R/T790M mutants through structural and energetic analysis. Int. J. Biol. Macromol. 2018, 118, 1948–1962. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and Biological Features Associated With Epidermal Growth Factor Receptor Gene Mutations in Lung Cancers. JNCI J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, Z.; Hao, X.; Hu, X.; Wang, H.; Wang, Y.; Ying, J. Clinical characteristics and response to tyrosine kinase inhibitors of patients with non-small cell lung cancer harboring uncommon epidermal growth factor receptor mutations. Chin. J. Cancer Res. 2017, 29, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Skaggs, B.J.; Gorre, M.E.; Ryvkin, A.; Burgess, M.R.; Xie, Y.; Han, Y.; Komisopoulou, E.; Brown, L.M.; Loo, J.A.; Landaw, E.M.; et al. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proc. Natl. Acad. Sci. USA 2006, 103, 19466–19471. [Google Scholar] [CrossRef] [Green Version]
- Sos, M.L.; Rode, H.B.; Heynck, S.; Peifer, M.; Fischer, F.; Klüter, S.; Pawar, V.G.; Reuter, C.; Heuckmann, J.M.; Weiss, J.; et al. Chemogenomic Profiling Provides Insights into the Limited Activity of Irreversible EGFR Inhibitors in Tumor Cells Expressing the T790M EGFR Resistance Mutation. Cancer Res. 2010, 70, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Kästner, J.; Senn, H.M.; Thiel, S.; Otte, N.; Thiel, W. QM/MM Free-Energy Perturbation Compared to Thermodynamic Integration and Umbrella Sampling: Application to an Enzymatic Reaction. J. Chem. Theory Comput. 2006, 2, 452–461. [Google Scholar] [CrossRef]
- Sutto, L.; Gervasio, F.L. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 10616–10621. [Google Scholar] [CrossRef] [Green Version]
- Gajiwala, K.S.; Feng, J.; Ferre, R.; Ryan, K.; Brodsky, O.; Weinrich, S.; Kath, J.C.; Stewart, A. Insights into the Aberrant Activity of Mutant EGFR Kinase Domain and Drug Recognition. Structure 2013, 21, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wykosky, J.; Fenton, T.; Furnari, F.; Cavenee, W.K. Therapeutic targeting of epidermal growth factor receptor in human cancer: Successes and limitations. Chin. J. Cancer 2011, 30, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Masood, A.; Kancha, R.K.; Subramanian, J. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non-small cell lung cancer harboring uncommon EGFR mutations: Focus on afatinib. Semin. Oncol. 2019, 46, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Yosaatmadja, Y.; Silva, S.; Dickson, J.M.; Patterson, A.V.; Smaill, J.B.; Flanagan, J.U.; McKeage, M.J.; Squire, C.J. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J. Struct. Biol. 2015, 192, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, M.; Liguori, A.; Karachaliou, N.; Gonzalez-Cao, M.; Daffinà, M.G.; D’Aveni, A.; Marabello, G.; Altavilla, G.; Rosell, R. Osimertinib in the treatment of non-small-cell lung cancer: Design, development and place in therapy. Lung Cancer Targets Ther. 2017, 8, 109–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liam, C.-K. The role of osimertinib in epidermal growth factor receptor (EGFR)-mutant non-small cell lung cancer. J. Thorac. Dis. 2019, 11, S448–S452. [Google Scholar] [CrossRef] [PubMed]
- Uchibori, K.; Inase, N.; Araki, M.; Kamada, M.; Sato, S.; Okuno, Y.; Fujita, N.; Katayama, R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017, 8, 14768. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhou, F.; Shen, W.; Jiang, T.; Wu, X.; Tong, X.; Shao, Y.W.; Qin, S.; Zhou, C. Novel Mutations on EGFR Leu792 Potentially Correlate to Acquired Resistance to Osimertinib in Advanced NSCLC. J. Thorac. Oncol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Klempner, S.J.; Mehta, P.; Schrock, A.B.; Ali, S.M.; Ou, S.-H.I. Cis-oriented solvent-front EGFR G796S mutation in tissue and ctDNA in a patient progressing on osimertinib: A case report and review of the literature. Lung Cancer 2017, 8, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Oztan, A.; Fischer, S.; Schrock, A.B.; Erlich, R.L.; Lovly, C.M.; Stephens, P.J.; Ross, J.S.; Miller, V.; Ali, S.M.; Ou, S.H.I.; et al. Emergence of EGFR G724S mutation in EGFR-mutant lung adenocarcinoma post progression on osimertinib. Lung Cancer 2017, 111. [Google Scholar] [CrossRef]
- Yella, J.; Yaddanapudi, S.; Wang, Y.; Jegga, A. Changing Trends in Computational Drug Repositioning. Pharmaceuticals 2018, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papapetropoulos, A.; Szabo, C. Inventing new therapies without reinventing the wheel: The power of drug repurposing. Br. J. Pharmacol. 2018, 175, 165–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, P.L.; John, T.; Dobrovic, A.; Mitchell, P. Prevalence and natural history of ALK positive non-small-cell lung cancer and the clinical impact of targeted therapy with ALK inhibitors. Clin. Epidemiol. 2014, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mano, H. ALKoma: A cancer subtype with a shared target. Cancer Discov. 2012, 2, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.; Kirstein, M.; Valentine, M.; Dittmer, K.; Shapiro, D.; Saltman, D.; Look, A. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef] [Green Version]
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Katayama, R. Drug resistance in anaplastic lymphoma kinase-rearranged lung cancer. Cancer Sci. 2018, 109, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Vernersson, E.; Khoo, N.K.S.; Henriksson, M.L.; Roos, G.; Palmer, R.H.; Hallberg, B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expr. Patterns 2006, 6, 448–461. [Google Scholar] [CrossRef]
- Pulford, K.; Lamant, L.; Morris, S.W.; Butler, L.H.; Wood, K.M.; Stroud, D.; Delsol, G.; Mason, D.Y. Detection of Anaplastic Lymphoma Kinase (ALK) and Nucleolar Protein Nucleophosmin (NPM)-ALK Proteins in Normal and Neoplastic Cells With the Monoclonal Antibody ALK1. Blood 1997, 89, 1394–1404. [Google Scholar] [CrossRef]
- Yao, S.; Cheng, M.; Zhang, Q.; Wasik, M.; Kelsh, R.; Winkler, C. Anaplastic Lymphoma Kinase Is Required for Neurogenesis in the Developing Central Nervous System of Zebrafish. PLoS ONE 2013, 8, e63757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrbough, J.; Broadie, K. Anterograde Jelly belly ligand to Alk receptor signaling at developing synapses is regulated by Mind the gap. Development 2010, 137, 3523–3533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klincumhom, N.; Tharasanit, T.; Thongkittidilok, C.; Tiptanavattana, N.; Rungarunlert, S.; Dinnyés, A.; Techakumphu, M. Selective TGF-β1/ALK inhibitor improves neuronal differentiation of mouse embryonic stem cells. Neurosci. Lett. 2014, 578, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Degoutin, J.; Brunet-de Carvalho, N.; Cifuentes-Diaz, C.; Vigny, M. ALK (Anaplastic Lymphoma Kinase) expression in DRG neurons and its involvement in neuron-Schwann cells interaction. Eur. J. Neurosci. 2009, 29, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Delisle, L.; Pierre-Eugène, C.; Bloch-Gallego, E.; Birling, M.-C.; Duband, J.-L.; Durand, E.; Bourgeois, T.; Matrot, B.; Sorg, T.; Huerre, M.; et al. Hyperactivation of Alk induces neonatal lethality in knock-in AlkF1178L mice. Oncotarget 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, R.; Lwigale, P. Expression of the heparin-binding growth factors Midkine and pleiotrophin during ocular development. Gene Expr. Patterns 2019, 32, 28–37. [Google Scholar] [CrossRef]
- Motegi, A. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J. Cell Sci. 2004, 117, 3319–3329. [Google Scholar] [CrossRef] [Green Version]
- Souttou, B.; Carvalho, N.B.-D.; Raulais, D.; Vigny, M. Activation of Anaplastic Lymphoma Kinase Receptor Tyrosine Kinase Induces Neuronal Differentiation through the Mitogen-activated Protein Kinase Pathway. J. Biol. Chem. 2001, 276, 9526–9531. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Jia, Y.; Li, N.; Sun, X.; Ng, K.; Ambing, E.; Gao, M.-Y.; Hua, S.; Chen, C.; Kim, S.; et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem. J. 2010, 430, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Pan, P.; Sun, H.; Xia, H.; Wang, X.; Li, Y.; Hou, T. Drug Discovery Targeting Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2019, 62, 10927–10954. [Google Scholar] [CrossRef]
- Holla, V.R.; Elamin, Y.Y.; Bailey, A.M.; Johnson, A.M.; Litzenburger, B.C.; Khotskaya, Y.B.; Sanchez, N.S.; Zeng, J.; Shufean, A.; Shaw, K.R.; et al. ALK: A tyrosine kinase target for cancer therapy. Mol. Case Stud. 2017, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27, iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Long, X.; Song, Y.; Chen, P.; Li, S.; Yang, H.; Wu, P.; Wang, Y.; Bing, Z.; Cao, Z.; et al. Distribution of ALK Fusion Variants and Correlation with Clinical Outcomes in Chinese Patients with Non-Small Cell Lung Cancer Treated with Crizotinib. Target. Oncol. 2019, 14, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.W.-S.; Leung, E.L.-H.; So, K.K.-T.; Tam, I.Y.-S.; Sihoe, A.D.-L.; Cheng, L.-C.; Ho, K.-K.; Au, J.S.-K.; Chung, L.-P.; Pik Wong, M. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009, 115, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Koivunen, J.P.; Mermel, C.; Zejnullahu, K.; Murphy, C.; Lifshits, E.; Holmes, A.J.; Choi, H.G.; Kim, J.; Chiang, D.; Thomas, R.; et al. EML4-ALK Fusion Gene and Efficacy of an ALK Kinase Inhibitor in Lung Cancer. Clin. Cancer Res. 2008, 14, 4275–4283. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Varghese, A.M.; Ou, S.-H.I.; Kabraji, S.; Awad, M.M.; Katayama, R.; Pawlak, A.; Mino-Kenudson, M.; Yeap, B.Y.; Riely, G.J.; et al. ALK Rearrangements Are Mutually Exclusive with Mutations in EGFR or KRAS: An Analysis of 1,683 Patients with Non-Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 4273–4281. [Google Scholar] [CrossRef] [Green Version]
- Tiseo, M.; Gelsomino, F.; Boggiani, D.; Bortesi, B.; Bartolotti, M.; Bozzetti, C.; Sammarelli, G.; Thai, E.; Ardizzoni, A. EGFR and EML4-ALK gene mutations in NSCLC: A case report of erlotinib-resistant patient with both concomitant mutations. Lung Cancer 2011, 71, 241–243. [Google Scholar] [CrossRef]
- Campos-Gomez, S.; Lara-Guerra, H.; Routbort, M.J.; Lu, X.; Simon, G.R. Lung adenocarcinoma with Concurrent KRAS Mutation and ALK Rearrangement Responding to Crizotinib: Case Report. Int. J. Biol. Markers 2015, 30, 254–257. [Google Scholar] [CrossRef]
- Bayliss, R.; Choi, J.; Fennell, D.A.; Fry, A.M.; Richards, M.W. Molecular mechanisms that underpin EML4-ALK driven cancers and their response to targeted drugs. Cell. Mol. Life Sci. 2016, 73, 1209–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, K.; Choi, Y.L.; Togashi, Y.; Soda, M.; Hatano, S.; Inamura, K.; Takada, S.; Ueno, T.; Yamashita, Y.; Satoh, Y.; et al. KIF5B-ALK, a Novel Fusion Oncokinase Identified by an Immunohistochemistry-based Diagnostic System for ALK-positive Lung Cancer. Clin. Cancer Res. 2009, 15, 3143–3149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.-H.I.; Klempner, S.J.; Greenbowe, J.R.; Azada, M.; Schrock, A.B.; Ali, S.M.; Ross, J.S.; Stephens, P.J.; Miller, V.A. Identification of a Novel HIP1-ALK Fusion Variant in Non–Small-Cell Lung Cancer (NSCLC) and Discovery of ALK I1171 (I1171N/S) Mutations in Two ALK-Rearranged NSCLC Patients with Resistance to Alectinib. J. Thorac. Oncol. 2014, 9, 1821–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.-L.; Lira, M.E.; Hong, M.; Kim, R.N.; Choi, S.-J.; Song, J.-Y.; Pandy, K.; Mann, D.L.; Stahl, J.A.; Peckham, H.E.; et al. A Novel Fusion of TPR and ALK in Lung Adenocarcinoma. J. Thorac. Oncol. 2014, 9, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, L.; Jiang, P.; Xu, F.; Zhang, W.; Guo, L.; Wu, J.; Zeng, Y.; Jiao, Y.; Ying, J. BIRC6-ALK, a Novel Fusion Gene in ALK Break-Apart FISH-Negative Lung Adenocarcinoma, Responds to Crizotinib. J. Thorac. Oncol. 2015, 10, e37–e39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Shao, Y.; Qin, H.-F.; Tai, Y.-H.; Gao, H.-J. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac. Cancer 2018, 9, 423–430. [Google Scholar] [CrossRef]
- Childress, M.A.; Himmelberg, S.M.; Chen, H.; Deng, W.; Davies, M.A.; Lovly, C.M. ALK Fusion Partners Impact Response to ALK Inhibition: Differential Effects on Sensitivity, Cellular Phenotypes, and Biochemical Properties. Mol. Cancer Res. 2018, 16, 1724–1736. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Zhang, Y.; Li, Y.; Liu, Q.; Chu, H.; Zhang, D.; Li, G. Integrating Multiple Accelerated Molecular Dynamics To Improve Accuracy of Free Energy Calculations. J. Chem. Theory Comput. 2018, 14, 1216–1227. [Google Scholar] [CrossRef]
- He, M.-Y.; Li, W.-K.; Zheng, Q.-C.; Zhang, H.-X. Conformational Transition of Key Structural Features Involved in Activation of ALK Induced by Two Neuroblastoma Mutations and ATP Binding: Insight from Accelerated Molecular Dynamics Simulations. ACS Chem. Neurosci. 2018, 9, 1783–1792. [Google Scholar] [CrossRef]
- Donella-Deana, A.; Marin, O.; Cesaro, L.; Gunby, R.H.; Ferrarese, A.; Coluccia, A.M.L.; Tartari, C.J.; Mologni, L.; Scapozza, L.; Gambacorti-Passerini, C.; et al. Unique Substrate Specificity of Anaplastic Lymphoma Kinase (ALK): Development of Phosphoacceptor Peptides for the Assay of ALK Activity. Biochemistry 2005, 44, 8533–8542. [Google Scholar] [CrossRef]
- Jiang, C.-H.; Huang, C.-X.; Chen, Y.-J.; Chuang, Y.-C.; Huang, B.-Y.; Yang, C.-N. Molecular Modeling for Structural Insights Concerning the Activation Mechanisms of F1174L and R1275Q Mutations on Anaplastic Lymphoma Kinase. Molecules 2018, 23, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Deng, R.; Jiang, H.; Song, H.; Li, S.; Shen, Q.; Huang, W.; Nussinov, R.; Yu, J.; Zhang, J. The Mechanism of ATP-Dependent Allosteric Protection of Akt Kinase Phosphorylation. Structure 2015, 23, 1725–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, M.; Gouaux, E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 2012, 485, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Li, S.; Zhang, J. Harnessing Allostery: A Novel Approach to Drug Discovery. Med. Res. Rev. 2014, 34, 1242–1285. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Huang, W.; Wang, Q.; Shen, Q.; Li, S.; Nussinov, R.; Zhang, J. The Structural Basis of ATP as an Allosteric Modulator. PLoS Comput. Biol. 2014, 10, e1003831. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in Advanced ALK -Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal–Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.-Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK Inhibitor Ceritinib Overcomes Crizotinib Resistance in Non-Small Cell Lung Cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Kay, M.; Dehghanian, F. Exploring the crizotinib resistance mechanism of NSCLC with the L1196M mutation using molecular dynamics simulation. J. Mol. Model. 2017, 23, 323. [Google Scholar] [CrossRef]
- Toyokawa, G.; Inamasu, E.; Shimamatsu, S.; Yoshida, T.; Nosaki, K.; Hirai, F.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Identification of a Novel ALK G1123S Mutation in a Patient with ALK-rearranged Non–small-cell Lung Cancer Exhibiting Resistance to Ceritinib. J. Thorac. Oncol. 2015, 10, e55–e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, X.; Niu, X.; Chang, L.; Chen, R.; Ou, S.-H.I.; Lu, S. Next generation sequencing reveals a novel ALK G1128A mutation resistant to crizotinib in an ALK-Rearranged NSCLC patient. Lung Cancer 2018, 123, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.C.; Dô, P.; Richard, N.; Dubos, C.; Michels, J.J.; Bonneau, J.; Gervais, R. Identification of I1171N resistance mutation in ALK-positive non-small-cell lung cancer tumor sample and circulating tumor DNA. Lung Cancer 2016, 99, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Yoda, S.; Lin, J.J.; Lawrence, M.S.; Burke, B.J.; Friboulet, L.; Langenbucher, A.; Dardaei, L.; Prutisto-Chang, K.; Dagogo-Jack, I.; Timofeevski, S.; et al. Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer. Cancer Discov. 2018, 8, 714–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.-H.I.; Greenbowe, J.; Khan, Z.U.; Azada, M.C.; Ross, J.S.; Stevens, P.J.; Ali, S.M.; Miller, V.A.; Gitlitz, B. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung Cancer 2015, 88, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.; Milliken, J.C.; Azada, M.C.; Miller, V.A.; Ali, S.M.; Klempner, S.J. ALK F1174V mutation confers sensitivity while ALK I1171 mutation confers resistance to alectinib. The importance of serial biopsy post progression. Lung Cancer 2016, 91, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two Novel ALK Mutations Mediate Acquired Resistance to the Next-Generation ALK Inhibitor Alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK Mutations in Lung Cancer That Confer Resistance to ALK Inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK -rearranged lung cancer. Cancer Discov. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.-W.; Lee, M.-S.; Lee, S.E.; Song, J.-Y.; Shin, H.-T.; Kim, Y.J.; Oh, D.Y.; Jung, K.; Sung, M.; Kim, M.; et al. Molecular breakdown: A comprehensive view of anaplastic lymphoma kinase ( ALK )-rearranged non-small cell lung cancer. J. Pathol. 2017, 243, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.Y.; Friboulet, L.; Kodack, D.P.; Engstrom, L.D.; Li, Q.; West, M.; Tang, R.W.; Wang, H.; Tsaparikos, K.; Wang, J.; et al. PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer Cell 2015, 28, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, Structure–Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro- N 2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)- N 4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,-4. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.-L.; Liu, W.; Dardaei, L.; et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuckmann, J.M.; Holzel, M.; Sos, M.L.; Heynck, S.; Balke-Want, H.; Koker, M.; Peifer, M.; Weiss, J.; Lovly, C.M.; Grutter, C.; et al. ALK Mutations Conferring Differential Resistance to Structurally Diverse ALK Inhibitors. Clin. Cancer Res. 2011, 17, 7394–7401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezquita, L.; Planchard, D. The role of brigatinib in crizotinib-resistant non-small cell lung cancer. Cancer Manag. Res. 2018, 10, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of Acquired Crizotinib Resistance in ALK-Rearranged Lung Cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovly, C.M.; Pao, W. Escaping ALK Inhibition: Mechanisms of and Strategies to Overcome Resistance. Sci. Transl. Med. 2012, 4, 120ps2. [Google Scholar] [CrossRef]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar] [CrossRef] [Green Version]
- Kodityal, S.; Elvin, J.A.; Squillace, R.; Agarwal, N.; Miller, V.A.; Ali, S.M.; Klempner, S.J.; Ou, S.-H.I. A novel acquired ALK F1245C mutation confers resistance to crizotinib in ALK-positive NSCLC but is sensitive to ceritinib. Lung Cancer 2016, 92, 19–21. [Google Scholar] [CrossRef]
- Michels, S.Y.F.; Scheel, A.H.; Wündisch, T.; Heuckmann, J.M.; Menon, R.; Puesken, M.; Kobe, C.; Pasternack, H.; Heydt, C.; Scheffler, M.; et al. ALK G1269A mutation as a potential mechanism of acquired resistance to crizotinib in an ALK-rearranged inflammatory myofibroblastic tumor. NPJ Precis. Oncol. 2017, 1, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.G.; Cortinovis, D.; Agustoni, F.; Arosio, G.; Villa, M.; Cordani, N.; Bidoli, P.; Bisson, W.H.; Pagni, F.; Piazza, R.; et al. A Compound L1196M/G1202R ALK Mutation in a Patient with ALK-Positive Lung Cancer with Acquired Resistance to Brigatinib Also Confers Primary Resistance to Lorlatinib. J. Thorac. Oncol. 2019, 14, e257–e259. [Google Scholar] [CrossRef] [PubMed]
- Recondo, G.; Mezquita, L.; Facchinetti, F.; Planchard, D.; Gazzah, A.; Bigot, L.; Rizvi, A.Z.; Frias, R.L.; Thiery, J.P.; Scoazec, J.-Y.; et al. Diverse Resistance Mechanisms to the Third-Generation ALK Inhibitor Lorlatinib in ALK-Rearranged Lung Cancer. Clin. Cancer Res. 2020, 26, 242–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Seto, Y.; Okada, K.; Uematsu, S.; Uchibori, K.; Tsukahara, M.; Oh-hara, T.; Fujita, N.; Yanagitani, N.; Nishio, M.; et al. Overcoming resistance by ALK compound mutation (I1171S + G1269A) after sequential treatment of multiple ALK inhibitors in non-small cell lung cancer. Thorac. Cancer 2020, 11, 581–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglivo, S.; Ricciuti, B.; Ludovini, V.; Metro, G.; Siggillino, A.; De Giglio, A.; Chiari, R. Dramatic Response to Lorlatinib in a Heavily Pretreated Lung Adenocarcinoma Patient Harboring G1202R Mutation and a Synchronous Novel R1192P ALK Point Mutation. J. Thorac. Oncol. 2018, 13, e145–e147. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; He, Z.; Xie, D.; Zheng, L.; Zhao, T.; Zhang, X.; Cheng, D. Molecular Mechanism Behind the Resistance of the G1202R-Mutated Anaplastic Lymphoma Kinase to the Approved Drug Ceritinib. J. Phys. Chem. B 2018, 122, 4680–4692. [Google Scholar] [CrossRef]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a Selective ALK Inhibitor Capable of Blocking the Resistant Gatekeeper Mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK -Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Kodama, T.; Hasegawa, M.; Takanashi, K.; Sakurai, Y.; Kondoh, O.; Sakamoto, H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother. Pharmacol. 2014, 74, 1023–1028. [Google Scholar] [CrossRef]
- Zhang, S.; Anjum, R.; Squillace, R.; Nadworny, S.; Zhou, T.; Keats, J.; Ning, Y.; Wardwell, S.D.; Miller, D.; Song, Y.; et al. The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clin. Cancer Res. 2016, 22, 5527–5538. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.W.; Richardson, P.F.; Bailey, S.; Brooun, A.; Burke, B.J.; Collins, M.R.; Cui, J.J.; Deal, J.G.; Deng, Y.-L.; Dinh, D.; et al. Discovery of (10 R )-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro- 2H -8,4-(metheno)pyrazolo[4,3- h ][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c. J. Med. Chem. 2014, 57, 4720–4744. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, R.; Wu, Y.; Song, M.; Li, J.; Yang, Q.; Chen, X.; Bao, J.; Zhao, Q. L1198F Mutation Resensitizes Crizotinib to ALK by Altering the Conformation of Inhibitor and ATP Binding Sites. Int. J. Mol. Sci. 2017, 18, 482. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Araki, M.; Sakashita, T.; Ma, B.; Kanada, R.; Yanagitani, N.; Horiike, A.; Koike, S.; Oh-hara, T.; Watanabe, K.; et al. Prediction of ALK mutations mediating ALK-TKIs resistance and drug re-purposing to overcome the resistance. EBioMedicine 2019, 41, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovly, C.M.; Heuckmann, J.M.; de Stanchina, E.; Chen, H.; Thomas, R.K.; Liang, C.; Pao, W. Insights into ALK-Driven Cancers Revealed through Development of Novel ALK Tyrosine Kinase Inhibitors. Cancer Res. 2011, 71, 4920–4931. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhou, J.; Zhou, J.; Feng, J.; Zhuang, W.; Chen, J.; Zhao, J.; Zhong, W.; Zhao, Y.; Zhang, Y.; et al. Efficacy, safety, and biomarker analysis of ensartinib in crizotinib-resistant, ALK-positive non-small-cell lung cancer: A multicentre, phase 2 trial. Lancet Respir. Med. 2020, 8, 45–53. [Google Scholar] [CrossRef]
- Horn, L.; Infante, J.R.; Reckamp, K.L.; Blumenschein, G.R.; Leal, T.A.; Waqar, S.N.; Gitlitz, B.J.; Sanborn, R.E.; Whisenant, J.G.; Du, L.; et al. Ensartinib (X-396) in ALK-Positive Non–Small Cell Lung Cancer: Results from a First-in-Human Phase I/II, Multicenter Study. Clin. Cancer Res. 2018, 24, 2771–2779. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Zhang, X.; Yang, D.; Wang, H.; Si, X.; Zhang, L. Single-center study to determine the safety and efficacy of CT-707 in Chinese patients with advanced anaplastic lymphoma kinase-rearranged non-small-cell lung cancer. Thorac. Cancer 2020, 11, 1216–1223. [Google Scholar] [CrossRef]
- Horn, L.; Reckamp, K.L.; Patel, S.; Blumenschein, G.; Neal, J.W.; Gitlitz, B.; Waqar, S.; Oxnard, G.; Brzezniak, C.; Dukart, G.; et al. Abstract CT151: CNS activity of ensartinib in ALK-positive non-small cell lung cancer patients. In Clinical Trials; American Association for Cancer Research: Philadelphia, PA, USA, 2017; p. CT151. [Google Scholar]
- Menichincheri, M.; Ardini, E.; Magnaghi, P.; Avanzi, N.; Banfi, P.; Bossi, R.; Buffa, L.; Canevari, G.; Ceriani, L.; Colombo, M.; et al. Discovery of Entrectinib: A New 3-Aminoindazole As a Potent Anaplastic Lymphoma Kinase (ALK), c-ros Oncogene 1 Kinase (ROS1), and Pan-Tropomyosin Receptor Kinases (Pan-TRKs) inhibitor. J. Med. Chem. 2016, 59, 3392–3408. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pizzutilo, E.G.; Marrapese, G.; Tosi, F.; Cerea, G.; Siena, S. Entrectinib for the treatment of metastatic NSCLC: Safety and efficacy. Expert Rev. Anticancer Ther. 2020, 20, 333–341. [Google Scholar] [CrossRef]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; De Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; et al. Entrectinib, a Pan-TRK, ROS1, and ALK Inhibitor with Activity in Multiple Molecularly Defined Cancer Indications. Mol. Cancer Ther. 2016, 15, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.; Kojima, H.; Saito, N.; Okabe, T.; Masuda, Y.; Furuya, T.; Nagano, T. Virtual screening and further development of novel ALK inhibitors. Bioorg. Med. Chem. 2011, 19, 3086–3095. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; Mcgrady, P.W.; et al. ALK Mutations Confer Differential Oncogenic Activation and Sensitivity to ALK Inhibition Therapy in Neuroblastoma. Cancer Cell 2014, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azam, M.; Seeliger, M.A.; Gray, N.S.; Kuriyan, J.; Daley, G.Q. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Biol. 2008, 15, 1109–1118. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, J.; Zhu, W. Mutation L1196M-induced conformational changes and the drug resistant mechanism of anaplastic lymphoma kinase studied by free energy perturbation and umbrella sampling. Phys. Chem. Chem. Phys. 2017, 19, 30239–30248. [Google Scholar] [CrossRef]
- Ai, X.; Shen, S.; Shen, L.; Lu, S. An interaction map of small-molecule kinase inhibitors with anaplastic lymphoma kinase (ALK) mutants in ALK-positive non-small cell lung cancer. Biochimie 2015, 112, 111–120. [Google Scholar] [CrossRef]
- Leonis, G.; Steinbrecher, T.; Papadopoulos, M.G. A Contribution to the Drug Resistance Mechanism of Darunavir, Amprenavir, Indinavir, and Saquinavir Complexes with HIV-1 Protease Due to Flap Mutation I50V: A Systematic MM–PBSA and Thermodynamic Integration Study. J. Chem. Inf. Model. 2013, 53, 2141–2153. [Google Scholar] [CrossRef]
- Dehghanian, F.; Kay, M.; Vallian, S. F1174V mutation alters the ALK active conformation in response to Crizotinib in NSCLC: Insight from molecular simulations. J. Mol. Graph. Model. 2017, 75, 287–293. [Google Scholar] [CrossRef]
- Liu, C.; Chen, C. P1.03-012 Using Computational Modeling to Simulate Clinical Response of ALK Inhibitors to G1202R ALK and Possible Mechanisms of Resistance. J. Thorac. Oncol. 2017, 12, S1955. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Guo, W.; Du, B.; Huang, X.; Wu, R.; Yang, B.; Lin, X.; Wu, Y. Insight into resistance mechanism of anaplastic lymphoma kinase to alectinib and JH-VIII-157-02 caused by G1202R solvent front mutation. Drug Des. Devel. Ther. 2018, 12, 1183–1193. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-Y.; Ji, F.-Q. A molecular dynamics investigation on the crizotinib resistance mechanism of C1156Y mutation in ALK. Biochem. Biophys. Res. Commun. 2012, 423, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Wang, X.; Zhang, T.; Jin, R.Z. Molecular dynamics simulations reveal the allosteric effect of F1174C resistance mutation to ceritinib in ALK-associated lung cancer. Comput. Biol. Chem. 2016, 65, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Nagasundaram, N.; Wilson Alphonse, C.R.; Samuel Gnana, P.V.; Rajaretinam, R.K. Molecular Dynamics Validation of Crizotinib Resistance to ALK Mutations (L1196M and G1269A) and Identification of Specific Inhibitors. J. Cell. Biochem. 2017, 118, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.-C.; Huang, B.-Y.; Chang, H.-W.; Yang, C.-N. Molecular Modeling of ALK L1198F and/or G1202R Mutations to Determine Differential Crizotinib Sensitivity. Sci. Rep. 2019, 9, 11390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhao, G.D.; Shi, Z.; Qi, L.L.; Zhou, L.Y.; Fu, Z.X. The Ras / Raf / MEK / ERK signaling pathway and its role in the occurrence and development of HCC ( Review ). Oncol. Lett. 2016, 3045–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, E.; Kondo, M. Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol. Res. 2011, 49, 248–268. [Google Scholar] [CrossRef]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef] [Green Version]
- Aramini, J.M.; Vorobiev, S.M.; Tuberty, L.M.; Janjua, H.; Campbell, E.T.; Seetharaman, J.; Su, M.; Huang, Y.J.; Acton, T.B.; Xiao, R.; et al. The RAS-Binding Domain of Human BRAF Protein Serine/Threonine Kinase Exhibits Allosteric Conformational Changes upon Binding HRAS. Structure 2015, 23, 1382–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutler, R.E.; Stephens, R.M.; Saracino, M.R.; Morrison, D.K. Autoregulation of the Raf-1 serine/threonine kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 9214–9219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef] [PubMed]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxnard, G.R.; Lo, P.C.; Nishino, M.; Dahlberg, S.E.; Lindeman, N.I.; Butaney, M.; Jackman, D.M.; Johnson, B.E.; Jänne, P.A. Natural history and molecular characteristics of lung cancers harboring egfr exon 20 insertions. J. Thorac. Oncol. 2013, 8, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.S.; Keshwani, M.M.; Steichen, J.M.; Kornev, A.P. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2517–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClendon, C.L.; Kornev, A.P.; Gilson, M.K.; Taylora, S.S. Dynamic architecture of a protein kinase. Proc. Natl. Acad. Sci. USA 2014, 111, E4623–E4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Gademann, K.; Portmann, C.; Blom, J.F.; Zeder, M.; Jüttner, F. Multiple toxin production in the cyanobacterium Microcystis: Isolation of the toxic protease inhibitor cyanopeptolin 1020. J. Nat. Prod. 2010, 73, 980–984. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and Role of Raf-1/B-Raf Heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.K.; Slupsky, J.R.; Andreas Kalmes, H.; Rapp, U.R. Active ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001, 61, 3595–3598. [Google Scholar] [PubMed]
- Haling, J.R.; Sudhamsu, J.; Yen, I.; Sideris, S.; Sandoval, W.; Phung, W.; Bravo, B.J.; Giannetti, A.M.; Peck, A.; Masselot, A.; et al. Structure of the BRAF-MEK Complex Reveals a Kinase Activity Independent Role for BRAF in MAPK Signaling. Cancer Cell 2014, 26, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Del Pino, G.G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF–MEK1–14-3-3 complexes. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. The importance of Raf dimerization in cell signaling. Small GTPases 2013, 4, 180. [Google Scholar] [CrossRef] [PubMed]
- Rajakulendran, T.; Sahmi, M.; Lefrançois, M.; Sicheri, F.; Therrien, M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 2009, 461, 542–545. [Google Scholar] [CrossRef]
- Karoulia, Z.; Wu, Y.; Ahmed, T.A.; Xin, Q.; Bollard, J.; Krepler, C.; Wu, X.; Zhang, C.; Bollag, G.; Herlyn, M.; et al. An Integrated Model of RAF Inhibitor Action Predicts Inhibitor Activity against Oncogenic BRAF Signaling. Cancer Cell 2016, 30, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Endicott, J.A.; Noble, M.E.M.; Johnson, L.N. The Structural Basis for Control of Eukaryotic Protein Kinases. Annu. Rev. Biochem. 2012, 81, 587–613. [Google Scholar] [CrossRef]
- Johnson, L.N.; Noble, M.E.M.; Owen, D.J. Active and inactive protein kinases: Structural basis for regulation. Cell 1996, 85, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cope, N.; Novak, B.; Candelora, C.; Wong, K.; Cavallo, M.; Gunderwala, A.; Liu, Z.; Li, Y.; Wang, Z. Biochemical Characterization of Full-Length Oncogenic BRAFV600E together with Molecular Dynamics Simulations Provide Insight into the Activation and Inhibition Mechanisms of RAF Kinases. ChemBioChem 2019, 20, 2850–2861. [Google Scholar] [CrossRef] [PubMed]
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for Carcinogenesis and Molecular Therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.H.; Zhou, H.; Zhu, S.B.; Huang, J.L.; Zhao, X.X.; Ding, H.; Pan, Y.L. Development of small-molecule therapeutics and strategies for targeting RAF kinase in BRAF-mutant colorectal cancer. Cancer Manag. Res. 2018, 10, 2289–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, S.M.; Carter, C.; Tang, L.Y.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Kane, R.C.; Farrell, A.T.; Saber, H.; Tang, S.; Williams, G.; Jee, J.M.; Liang, C.; Booth, B.; Chidambaram, N.; Morse, D.; et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 2006, 12, 7271–7278. [Google Scholar] [CrossRef] [Green Version]
- Lang, L. FDA Approves Sorafenib for Patients With Inoperable Liver Cancer. Gastroenterology 2008, 134, 379. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I dose-escalation and -expansion study of the BRAF inhibitor encorafenib (LGX818) in metastatic BRAF-mutant melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, D.D.; Li, N.; Poon, D.J.; Aardalen, K.; Kaufman, S.; Merritt, H.; Salangsang, F.; Lorenzana, E.; Li, A.; Ghoddusi, M.; et al. Abstract 3790: Preclinical profile of LGX818: A potent and selective RAF kinase inhibitor. Cancer Res. 2012, 72, 3790. [Google Scholar] [CrossRef]
- Thevakumaran, N.; Lavoie, H.; Critton, D.A.; Tebben, A.; Marinier, A.; Sicheri, F.; Therrien, M. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat. Struct. Mol. Biol. 2014, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Zhang, Y.; van Horn, R.D.; Yin, T.; Buchanan, S.; Yadav, V.; Mochalkin, I.; Wong, S.S.; Yue, Y.G.; Huber, L.; et al. Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov. 2016, 6, 300–315. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, H.; Thevakumaran, N.; Gavory, G.; Li, J.J.; Padeganeh, A.; Guiral, S.; Duchaine, J.; Mao, D.Y.L.; Bouvier, M.; Sicheri, F.; et al. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat. Chem. Biol. 2013, 9, 428–436. [Google Scholar] [CrossRef]
- Tse, A.; Verkhivker, G.M. Exploring Molecular Mechanisms of Paradoxical Activation in the BRAF Kinase Dimers: Atomistic Simulations of Conformational Dynamics and Modeling of Allosteric Communication Networks and Signaling Pathways. PLoS ONE 2016, 11, e0166583. [Google Scholar] [CrossRef]
- Waizenegger, I.C.; Baum, A.; Steurer, S.; Stadtmüller, H.; Bader, G.; Schaaf, O.; Garin-Chesa, P.; Schlattl, A.; Schweifer, N.; Haslinger, C.; et al. A novel RAF kinase inhibitor with DFG-out-binding mode: High efficacy in BRAF-mutant tumor xenograft models in the absence of normal tissue hyperproliferation. Mol. Cancer Ther. 2016, 15, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.B.; Henry, J.R.; Kaufman, M.D.; Lu, W.P.; Smith, B.D.; Vogeti, S.; Rutkoski, T.J.; Wise, S.; Chun, L.; Zhang, Y.; et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015, 28, 384–398. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Di Michele, M.; Stes, E.; Vandermarliere, E.; Martens, L.; Gevaert, K.; Van Heerde, E.; Linders, J.T.M.; Brehmer, D.; Jacoby, E.; et al. Structural investigation of B-Raf paradox breaker and inducer inhibitors. J. Med. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Noeparast, A.; Verschelden, G.; Umelo, I.; de Brakeleer, S.; Decoste, L.; Teugels, E.; de Greve, J. Investigation of non-V600 BRAF mutations commonly found in NSCLC for their sensitivity to Dabrafenib or Trametinib. Ann. Oncol. 2015, 26, ii36. [Google Scholar] [CrossRef]
- Mazieres, J.; Cropet, C.; Montané, L.; Barlesi, F.; Souquet, P.J.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in non-small-cell lung cancer patients with BRAFV600 and BRAFnonV600 mutations. Ann. Oncol. 2020, 31, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Casadei Gardini, A.; Chiadini, E.; Faloppi, L.; Marisi, G.; Delmonte, A.; Scartozzi, M.; Loretelli, C.; Lucchesi, A.; Oboldi, D.; Dubini, A.; et al. Efficacy of sorafenib in BRAF-mutated nonsmall-cell lung cancer (NSCLC) and no response in synchronous BRAF wild typehepatocellular carcinoma: A case report. BMC Cancer 2016, 16. [Google Scholar] [CrossRef]
- Sereno, M.; Moreno, V.; Rubio, J.M.; Gómez-Raposo, C.; Sánchez, S.G.; Jusdado, R.H.; Falagan, S.; Tébar, F.Z.; Sáenz, E.C. A significant response to sorafenib in a woman with advanced lung adenocarcinoma and a BRAF non-V600 mutation. Anticancer. Drugs 2015, 26, 1004–1007. [Google Scholar] [CrossRef]
- Gautschi, O.; Peters, S.; Zoete, V.; Aebersold-Keller, F.; Strobel, K.; Schwizer, B.; Hirschmann, A.; Michielin, O.; Diebold, J. Lung adenocarcinoma with BRAF G469L mutation refractory to vemurafenib. Lung Cancer 2013, 82, 365–367. [Google Scholar] [CrossRef]
- Hallmeyer, S.; Gonzalez, R.; Lawson, D.H.; Cranmer, L.D.; Linette, G.P.; Puzanov, I.; Taback, B.; Cowey, C.L.; Ribas, A.; Daniels, G.A.; et al. Vemurafenib treatment for patients with locally advanced, unresectable stage IIIC or metastatic melanoma and activating exon 15 BRAF mutations other than V600E. Melanoma Res. 2017, 27, 585–590. [Google Scholar] [CrossRef]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Gadaleta-Caldarola, G.; Divella, R.; Mazzocca, A.; Infusino, S.; Ferraro, E.; Filippelli, G.; Daniele, A.; Sabbà, C.; Abbate, I.; Brandi, M. Sorafenib: The gold standard therapy in advanced hepatocellular carcinoma and beyond. Futur. Oncol. 2015, 11, 2263–2266. [Google Scholar] [CrossRef]
- Rubinstein, J.C.; Sznol, M.; Pavlick, A.C.; Ariyan, S.; Cheng, E.; Bacchiocchi, A.; Kluger, H.M.; Narayan, D.; Halaban, R. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J. Transl. Med. 2010, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, O.; Clements, A.; Menzies, A.M.; O’Toole, S.; Kefford, R.F.; Long, G.V. BRAF inhibitor activity in V600R metastatic melanoma. Eur. J. Cancer 2013, 49, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Moiseyenko, F.V.; Egorenkov, V.V.; Kramchaninov, M.M.; Artemieva, E.V.; Aleksakhina, S.N.; Holmatov, M.M.; Moiseyenko, V.M.; Imyanitov, E.N. Lack of Response to Vemurafenib in Melanoma Carrying BRAF K601E Mutation. Case Rep. Oncol. 2019, 12, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Landrette, S.F.; Wang, T.; Evans, P.; Bacchiocchi, A.; Bjornson, R.; Cheng, E.; Stiegler, A.L.; Gathiaka, S.; Acevedo, O.; et al. Identification of PLX4032-resistance mechanisms and implications for novel RAF inhibitors. Pigment Cell Melanoma Res. 2014, 27, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yao, Z.; Jonsson, P.; Allen, A.N.; Qin, A.C.R.; Uddin, S.; Dunkel, I.J.; Petriccione, M.; Manova, K.; Haque, S.; et al. A secondary mutation in BRAF confers resistance to RAF inhibition in a BRAFV600E-mutant brain tumor. Cancer Discov. 2018, 8, 1130–1141. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.; Kirk, R.; Hayward, R.; Zambon, A.; Viros, A.; Cantarino, N.; Affolter, A.; Nourry, A.; Niculescu-Duvaz, D.; Springer, C.; et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci. Transl. Med. 2010, 2, 35ra41. [Google Scholar] [CrossRef]
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S.F. The ras superfamily of small GTPases: The unlocked secrets. Methods Mol. Biol. 2014, 1120, 1–18. [Google Scholar]
- Liao, J.; Shima, F.; Araki, M.; Ye, M.; Muraoka, S.; Sugimoto, T.; Kawamura, M.; Yamamoto, N.; Tamura, A.; Kataoka, T. Two conformational states of Ras GTPase exhibit differential GTP-binding kinetics. Biochem. Biophys. Res. Commun. 2008, 369, 327–332. [Google Scholar] [CrossRef]
- Hennig, A.; Markwart, R.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Ras activation revisited: Role of GEF and GAP systems. Biol. Chem. 2015, 396, 831–848. [Google Scholar] [CrossRef] [PubMed]
- Dreusicke, D.; Schulz, G.E. The glycine-rich loop of adenylate kinase forms a giant anion hole. FEBS Lett. 1986, 208, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Klockow, B.; Ahmadian, M.R.; Block, C.; Wittinghofer, A. Oncogenic insertional mutations in the P-loop of Ras are overactive in MAP kinase signaling. Oncogene 2000, 19, 5367–5376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, A.; Travesset, A. Differential dynamics of RAS isoforms in GDP- and GTP-bound states. Proteins Struct. Funct. Bioinforma. 2015, 83, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Prakash, P.; Sayyed-Ahmad, A.; Gorfe, A.A. pMD-Membrane: A Method for Ligand Binding Site Identification in Membrane-Bound Proteins. PLOS Comput. Biol. 2015, 11, e1004469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayyed-Ahmad, A.; Gorfe, A.A. Mixed-Probe Simulation and Probe-Derived Surface Topography Map Analysis for Ligand Binding Site Identification. J. Chem. Theory Comput. 2017, 13, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.N.; Bellamacina, C.R.; Ding, X.; Jeffery, C.J.; Mattos, C.; Petsko, G.A.; Ringe, D. An experimental approach to mapping the binding surfaces of crystalline proteins. J. Phys. Chem. 1996, 100, 2605–2611. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Huth, J.R.; Fesik, S.W. Druggability indices for protein targets derived from NMr-based screening data. J. Med. Chem. 2005, 48, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Hocker, H.J.; Cho, K.J.; Chen, C.Y.K.; Rambahal, N.; Sagineedu, S.R.; Shaari, K.; Stanslas, J.; Hancock, J.F.; Gorfe, A.A. Andrographolide derivatives inhibit guanine nucleotide exchange and abrogate oncogenic Ras function. Proc. Natl. Acad. Sci. USA 2013, 110, 10201–10206. [Google Scholar] [CrossRef] [Green Version]
- Rosnizeck, I.C.; Graf, T.; Spoerner, M.; Tränkle, J.; Filchtinski, D.; Herrmann, C.; Gremer, L.; Vetter, I.R.; Wittinghofer, A.; König, B.; et al. Stabilizing a weak binding state for effectors in the human ras protein by cyclen complexes. Angew. Chem. Int. Ed. 2010, 49, 3830–3833. [Google Scholar] [CrossRef]
- Prakash, P.; Hancock, J.F.; Gorfe, A.A. Binding hotspots on K-ras: Consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins Struct. Funct. Bioinforma. 2015, 83, 898–909. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.H.; Wellen, S.L.; Klimstra, D.; Lenczowski, J.M.; Tichelaar, J.W.; Lizak, M.J.; Whitsett, J.A.; Koretsky, A.; Varmus, H.E. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001, 15, 3249–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Alexander, R.E.; Maclennan, G.T.; Cummings, O.W.; Montironi, R.; Lopez-beltran, A.; Cramer, H.M.; Davidson, D.D. Molecular pathology of lung cancer: Key to personalized medicine. Mod. Pathol. 2012, 25, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Marín-Ramos, N.I.; Piñar, C.; Vázquez-Villa, H.; Martín-Fontecha, M.; González, Á.; Canales, Á.; Algar, S.; Mayo, P.P.; Jiménez-Barbero, J.; Gajate, C.; et al. Development of a Nucleotide Exchange Inhibitor That Impairs Ras Oncogenic Signaling. Chem. A Eur. J. 2017, 23, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patgiri, A.; Yadav, K.K.; Arora, P.S.; Bar-Sagi, D. An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 2011, 7, 585–587. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Burke, J.P.; Phan, J.; Burns, M.C.; Olejniczak, E.T.; Waterson, A.G.; Lee, T.; Rossanese, O.W.; Fesik, S.W. Discovery of Small Molecules that Bind to K-Ras and Inhibit Sos-Mediated Activation. Angew. Chem. Int. Ed. 2012, 124, 6244–6247. [Google Scholar] [CrossRef] [Green Version]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D.; et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef] [Green Version]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [Green Version]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular Epidemiology of EGFR and KRAS Mutations in 3,026 Lung Adenocarcinomas: Higher Susceptibility of Women to Smoking-Related KRAS-Mutant Cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [Green Version]
- El Osta, B.E.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and outcomes of patients (pts) with metastatic KRAS mutant lung adenocarcinomas: Lung Cancer Mutation Consortium (LCMC) database. J. Clin. Oncol. 2017, 35, 9021. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Domerg, C.; Hainaut, P.; Jänne, P.A.; Pignon, J.-P.; Graziano, S.; Douillard, J.-Y.; Brambilla, E.; Le Chevalier, T.; Seymour, L.; et al. Pooled Analysis of the Prognostic and Predictive Effects of KRAS Mutation Status and KRAS Mutation Subtype in Early-Stage Resected Non–Small-Cell Lung Cancer in Four Trials of Adjuvant Chemotherapy. J. Clin. Oncol. 2013, 31, 2173–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aredo, J.V.; Padda, S.K.; Kunder, C.A.; Han, S.S.; Neal, J.W.; Shrager, J.B.; Wakelee, H.A. Impact of KRAS mutation subtype and concurrent pathogenic mutations on non-small cell lung cancer outcomes. Lung Cancer 2019, 133, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Bera, A.K.; Gondi, S.; Westover, K.D. KRAS Switch Mutants D33E and A59G Crystallize in the State 1 Conformation. Biochemistry 2018, 57, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-C.; Er, T.-K.; Liu, Y.-Y.; Hwang, J.-K.; Barrio, M.J.; Rodrigo, M.; Garcia-Toro, E.; Herreros-Villanueva, M. Computational Analysis of KRAS Mutations: Implications for Different Effects on the KRAS p.G12D and p.G13D Mutations. PLoS ONE 2013, 8, e55793. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.A.; Volmar, A.Y.; Pavlopoulos, S.; Mattos, C. K-Ras Populates Conformational States Differently from Its Isoform H-Ras and Oncogenic Mutant K-RasG12D. Structure 2018, 26, 810–820.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Long, B.N.; Boris, G.H.; Chen, A.; Ni, S.; Kennedy, M.A. Structural insight into the rearrangement of the switch I region in GTP-bound G12A K-Ras. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 970–984. [Google Scholar] [CrossRef]
- Vatansever, S.; Erman, B.; Gümüş, Z.H. Oncogenic G12D mutation alters local conformations and dynamics of K-Ras. Sci. Rep. 2019, 9, 11730. [Google Scholar] [CrossRef] [Green Version]
- Kauke, M.J.; Traxlmayr, M.W.; Parker, J.A.; Kiefer, J.D.; Knihtila, R.; McGee, J.; Verdine, G.; Mattos, C.; Wittrup, K.D. An engineered protein antagonist of K-Ras/B-Raf interaction. Sci. Rep. 2017, 7, 5831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.C.; Gurbani, D.; Ficarro, S.B.; Carrasco, M.A.; Lim, S.M.; Choi, H.G.; Xie, T.; Marto, J.A.; Chen, Z.; Gray, N.S.; et al. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. USA 2014, 111, 8895–8900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.M.; Westover, K.D.; Ficarro, S.B.; Harrison, R.A.; Choi, H.G.; Pacold, M.E.; Carrasco, M.; Hunter, J.; Kim, N.D.; Xie, T.; et al. Therapeutic targeting of oncogenic K-ras by a covalent catalytic site inhibitor. Angew. Chem. Int. Ed. 2014, 53, 199–204. [Google Scholar] [CrossRef]
- Sun, Q.; Phan, J.; Friberg, A.R.; Camper, D.V.; Olejniczak, E.T.; Fesik, S.W. A method for the second-site screening of K-Ras in the presence of a covalently attached first-site ligand. J. Biomol. NMR 2014, 60, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Migoni, A.; Canning, P.; Quevedo, C.E.; Bataille, C.J.R.; Bery, N.; Miller, A.; Russell, A.J.; Phillips, S.E.V.; Carr, S.B.; Rabbitts, T.H. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc. Natl. Acad. Sci. USA 2019, 116, 2545–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Gupta, A.K.; Wang, X.; Pagba, C.V.; Prakash, P.; Sarkar-Banerjee, S.; Putkey, J.; Gorfe, A.A. Multi-target, ensemble-based virtual screening yields novel allosteric KRAS inhibitors at high success rate. Chem. Biol. Drug Des. 2019, cbdd.13519. [Google Scholar] [CrossRef]
- Putora, P.M.; Ess, S.; Panje, C.; Hundsberger, T.; van Leyen, K.; Plasswilm, L.; Früh, M. Prognostic significance of histology after resection of brain metastases and whole brain radiotherapy in non-small cell lung cancer (NSCLC). Clin. Exp. Metastasis 2015, 32, 143–149. [Google Scholar] [CrossRef]
- Su, S.F.; Hu, Y.X.; Ouyang, W.W.; Ma, Z.; Lu, B.; Li, Q.S.; Li, H.Q.; Wang, Z.Y.; Wang, Y. The survival outcomes and prognosis of stage IV non-small-cell lung cancer treated with thoracic three-dimensional radiotherapy combined with chemotherapy. Radiat. Oncol. 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Guise, J.-M.; Savitz, L.A.; Friedman, C.P. Mind the Gap: Putting Evidence into Practice in the Era of Learning Health Systems. J. Gen. Intern. Med. 2018, 33, 2237–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, O.B.; Neves, K.; Wasilewska-Sampaio, A.P.; Carneiro, C.F. The Brazilian Reproducibility Initiative. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-C. Beware of Docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, D.L.; van Herpen, C.M.L.; van Laarhoven, H.W.M.; Smit, E.F.; Groen, H.J.M.; Willems, S.M.; Nederlof, P.M.; Langenberg, M.H.G.; Cuppen, E.; Sleijfer, S.; et al. Molecular Tumor Boards: Current practice and future needs. Ann. Oncol. 2017, 28, 3070–3075. [Google Scholar] [CrossRef]
- Santos, J.F.V.M.R.; Hiddinga, B.I.; van Kempen, L.C.; ter Elst, A.; Hijmering, L.B.; Hiltermann, T.J.; Schuuring, E.; Groves, M.R.; Groen, H.J.; Wekken, A.J. van der Abstract 1398: Modeling of drug-protein interactions to support clinical decision making for therapy-resistant EGFR or ALK -positive non-small cell lung carcinoma. In Clinical Research (Excluding Clinical Trials); American Association for Cancer Research: Philadelphia, PA, USA, 2019; Volume 79, p. 1398. [Google Scholar]
- Koopman, B.; van der Wekken, A.J.; ter Elst, A.; Hiltermann, T.J.N.; Vilacha, J.F.; Groves, M.R.; van den Berg, A.; Hiddinga, B.I.; Hijmering-Kappelle, L.B.M.; Stigt, J.A.; et al. Relevance and Effectiveness of Molecular Tumor Board Recommendations for Patients With Non–Small-Cell Lung Cancer With Rare or Complex Mutational Profiles. JCO Precis. Oncol. 2020, 393–410. [Google Scholar] [CrossRef]












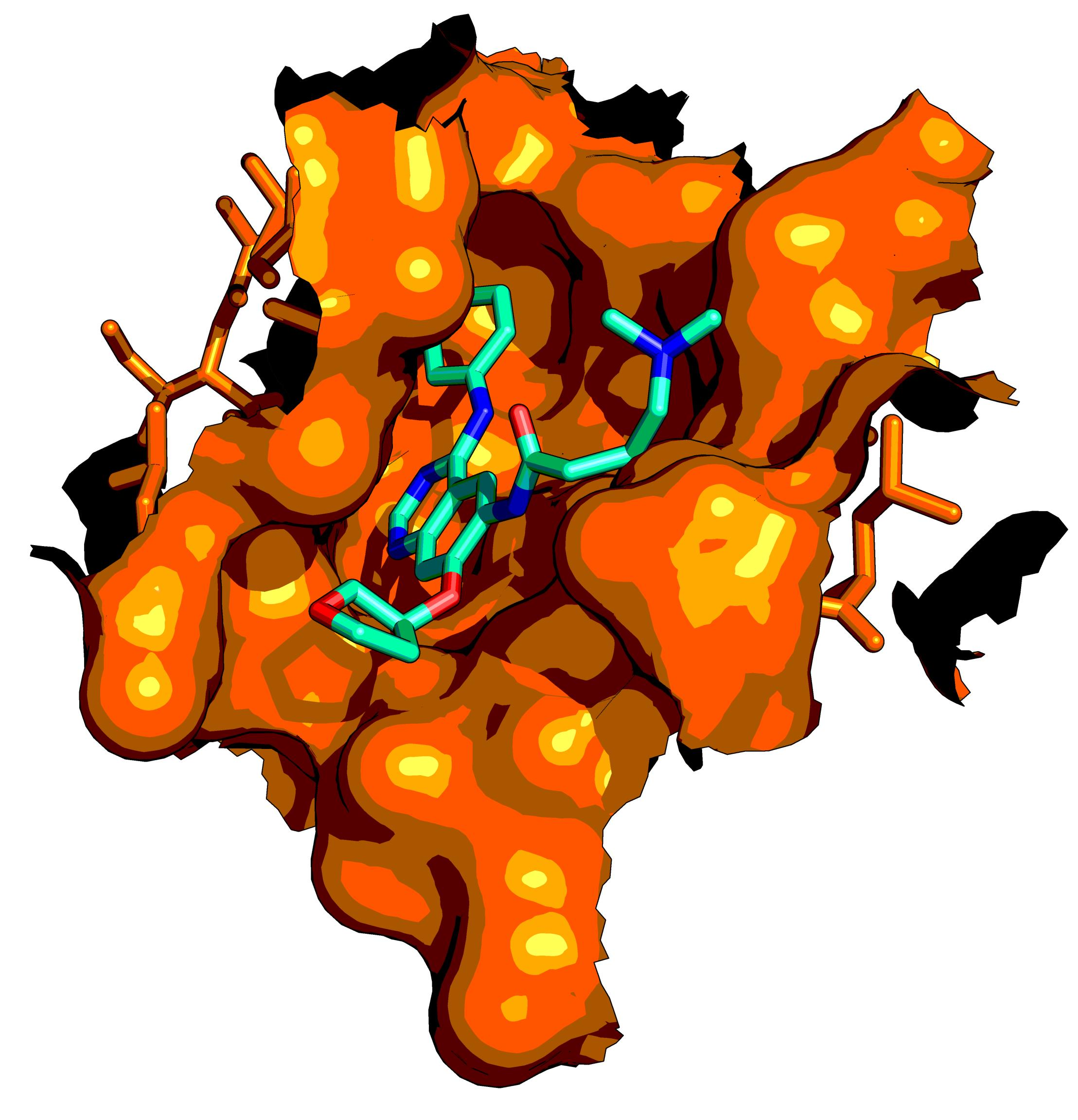{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutations | 1st Generation | 2nd Generation | 3rd Generation | ||
|---|---|---|---|---|---|
| Erlotinib | Gefitinib | Afatinib | Dacomitinib | Osimertinib | |
| L718V/Q | Resistant [63] | Resistant [64] | N/A | N/A | Resistant [64] |
| G719A | N/A | N/A | Sensitive [59] | N/A | N/A |
| G719S | Sensitive [65] | Sensitive [44,65,66,67] | N/A | N/A | N/A |
| G724S | N/A | N/A | Sensitive [68] | N/A | Resistant [68] |
| T790M | Resistant [44,50] | Resistant [44] | N/A | N/A | Sensitive [59,69] |
| L792H/F/Y | N/A | Resistant [64] | N/A | N/A | Resistant [64] |
| G796C | Resistant [63] | N/A | N/A | N/A | Resistant [70] |
| G796D | Resistant [63] | N/A | N/A | N/A | Resistant [71] |
| G796R | Resistant [63] | N/A | N/A | N/A | Resistant [64] |
| G796S | N/A | N/A | N/A | N/A | Resistant [72] |
| C797S | Resistant [73] | Sensitive [64] | Resistant [63] | N/A | Resistant [74] |
| L858R | Sensitive [60,65,75,76] | Sensitive [60,65,75,76] | Sensitive [59,77] | Sensitive [78] | Sensitive [69,79] |
| L858R/L718V | N/A | N/A | Sensitive [80] | N/A | Resistant [80] |
| L858R/L718Q | N/A | Resistant [64] | Sensitive [81] | N/A | Resistant [64,81] |
| L858R/G724S | N/A | N/A | N/A | N/A | Resistant [82] |
| L858R/T790M | Resistant [50,59,60,65,77,78] | Resistant [59,60,65,67,77] | Inconclusive [77] | Resistant [78] | Sensitive [78,79,83] |
| L858R/C797S | Sensitive [78] | Sensitive [78] | Sensitive [78] | N/A | Resistant [84] |
| T790M/G719S | N/A | Resistant [48,65] | N/A | N/A | N/A |
| T790M/C797S | Resistant [63,83] | Resistant [83] | Resistant [83] | Resistant [83] | Resistant [59,83] |
| L858R/ T790M/ C797S | Resistant [78] | N/A | Resistant [78] | Resistant [78] | Resistant [78] |
| Exon19del* | Sensitive [66,75,76] | Sensitive [66] | Sensitive [85,86] | Sensitive [78] | Sensitive [64] |
| Exon19del*/ G724S | N/A | N/A | Sensitive [68] | N/A | Resistant [68] |
| Exon19del* / T790M | Resistant [65] | Resistant [65] | N/A | Resistant [74] | Sensitive [78] |
| Exon19del*/ C797S | N/A | Sensitive [74] | Sensitive [74] | N/A | Resistant [64,84] |
| L858R/T790M/ L718Q | Resistant [81] | Resistant [81] | Resistant [81] | Resistant [81] | Resistant [81] |
| Exon19del*/ T790M/ P794L | N/A | N/A | Sensitive [87] | N/A | Resistant [87] |
| Exon19del*/ T790M/ C797S | Resistant [74] | Resistant [74] | Resistant [74] | Resistant [74] | Resistant [74] |
| Mutations | 1st Generation | 2nd Generation | 3rd Generation | |||
|---|---|---|---|---|---|---|
| Crizotinib | Entrectinib * | Alectinib | Brigatinib | Ceritinib | Lorlatinib | |
| G1123S | Resistant [164] | N/A | N/A | N/A | Resistant [164] | N/A |
| G1128A | Resistant [165] | N/A | N/A | N/A | N/A | N/A |
| I1171N | N/A | N/A | Resistant [146,166,167] | N/A | Sensitive [168] | Sensitive [167] |
| I1171S | N/A | N/A | Resistant [146,169] | N/A | N/A | N/A |
| I1171T | Resistant [170] | N/A | Resistant [170] | N/A | Sensitive [162] | N/A |
| C1156Y | Resistant [171] | N/A | N/A | N/A | N/A | N/A |
| F1174C | Resistant [162,170,172] | N/A | Sensitive [162,170,172] | N/A | Resistant [162,170,172] | N/A |
| F1174L | N/A | N/A | Sensitive [162,170,172] | N/A | Resistant [162,170,172] | N/A |
| F1174V | Resistant [169] | N/A | Sensitive [162,169,170,172] | N/A | Resistant [162,170,172] | N/A |
| V1180L | Resistant [170] | N/A | Resistant [170] | N/A | Sensitive [170] | N/A |
| R1192P | Resistant [173] | N/A | N/A | N/A | Resistant [173] | N/A |
| L1196M | N/A | N/A | N/A | Sensitive [174] | Sensitive [162,175] | Sensitive [174] |
| L1196Q | N/A | N/A | Sensitive [167] | N/A | N/A | N/A |
| L1198F | Sensitive [176] | N/A | N/A | N/A | N/A | Resistant [176] |
| L1198P | Resistant [177] | N/A | N/A | N/A | N/A | N/A |
| G1202R | Resistant [162,172] | N/A | Resistant [167,170,172] | Sensitive [178] | Resistant [162,172] | Sensitive [167,179] |
| D1203N | Resistant [177] | N/A | N/A | N/A | N/A | N/A |
| S1206Y | Resistant [180,181,182] | N/A | N/A | N/A | N/A | N/A |
| E1210K | N/A | N/A | N/A | N/A | N/A | Resistant [167] |
| F1245C | Resistant [183] | N/A | N/A | N/A | Sensitive [183] | N/A |
| G1269A | Resistant [184] | N/A | Resistant [180,181,182] | N/A | Sensitive [162,175] | Resistant [167] |
| A1280V | Resistant [173] | N/A | N/A | N/A | Resistant [173] | N/A |
| L1535Q | Resistant [173] | N/A | N/A | N/A | Resistant [173] | N/A |
| E1210K/D1203N | Resistant [172] | N/A | Resistant [172] | Resistant [172] | Resistant [172] | Sensitive [172] |
| C1156Y/L1198F | N/A | N/A | N/A | N/A | N/A | Resistant [167] |
| D1203N/E1210K | Resistant [172] | N/A | Resistant [172] | Resistant [172] | Resistant [172] | Sensitive [172] |
| D1203N/F1174C | Resistant [172] | N/A | Resistant [172] | Resistant [172] | Resistant [172] | Sensitive [172] |
| L1196M/G1202R | N/A | N/A | N/A | Resistant [185] | N/A | Resistant [185] |
| L1196M/D1203N | N/A | N/A | N/A | N/A | N/A | Resistant [186] |
| I1171S/G1269A | N/A | N/A | N/A | Sensitive [187] | Sensitive [187] | Resistant [187] |
| C1156Y/G169A | Resistant [186] | Resistant [186] | Resistant [186] | N/A | N/A | Sensitive [186] |
| F1174L/G1202R | N/A | N/A | N/A | N/A | N/A | Resistant [186] |
| G1202R/R1192P | Resistant [188] | N/A | N/A | Resistant [188] | N/A | Sensitive [188] |
| Mutations | 1st Generation | 2nd Generation | ||
|---|---|---|---|---|
| Sorafenib | Dabrafenib | Encorafenib | Vemurafenib | |
| G466A | N/A | Sensitive [267] | N/A | Resistant [268] |
| G466V | N/A | Sensitive [267] | N/A | Resistant [268] |
| G469A | N/A | Sensitive [267] | N/A | Resistant [268] |
| G469V | Sensitive [269] | Sensitive [267] | N/A | Resistant [268] |
| G469R | Sensitive [270] | N/A | N/A | N/A |
| G469L | N/A | N/A | N/A | Resistant [271] |
| D594E | N/A | Sensitive [267] | N/A | N/A |
| D594G | N/A | N/A | N/A | Sensitive [272] |
| D594N | N/A | Sensitive [267] | N/A | N/A |
| G596C | N/A | Sensitive [267] | N/A | N/A |
| G596R | N/A | N/A | N/A | Resistant [268] |
| L597S | N/A | N/A | N/A | Sensitive [272] |
| V600D | N/A | Sensitive [273] | N/A | Resistant [268] |
| V600E | Inconclusive ** [269,274] | Sensitive [253] | N/A | Sensitive [253] |
| V600K | N/A | Sensitive [275] | N/A | Sensitive [275] |
| V600M | N/A | Sensitive [273] | N/A | N/A |
| V600R | N/A | Sensitive [276] | N/A | Sensitive [276] |
| K601E | N/A | N/A | N/A | Resistant [277] |
| V600E_L505H | N/A | N/A | N/A | Resistant [278] |
| V600E_L514V | N/A | Resistant [279] | N/A | Resistant [279] |
| V600E_T529I | Resistant [280] | N/A | N/A | N/A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilachã, J.F.; Mitchel, S.C.; Akele, M.Z.; Evans, S.; Groves, M.R. Making NSCLC Crystal Clear: How Kinase Structures Revolutionized Lung Cancer Treatment. Crystals 2020, 10, 725. https://doi.org/10.3390/cryst10090725
Vilachã JF, Mitchel SC, Akele MZ, Evans S, Groves MR. Making NSCLC Crystal Clear: How Kinase Structures Revolutionized Lung Cancer Treatment. Crystals. 2020; 10(9):725. https://doi.org/10.3390/cryst10090725
Chicago/Turabian StyleVilachã, Juliana F., Sarah C. Mitchel, Muluembet Z. Akele, Stephen Evans, and Matthew R. Groves. 2020. "Making NSCLC Crystal Clear: How Kinase Structures Revolutionized Lung Cancer Treatment" Crystals 10, no. 9: 725. https://doi.org/10.3390/cryst10090725
APA StyleVilachã, J. F., Mitchel, S. C., Akele, M. Z., Evans, S., & Groves, M. R. (2020). Making NSCLC Crystal Clear: How Kinase Structures Revolutionized Lung Cancer Treatment. Crystals, 10(9), 725. https://doi.org/10.3390/cryst10090725