3.1. Ab Initio Calculations of ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 Bulk Properties
In order to begin the calculations, by means of the B3LYP or B3PW functional, the ReO
3, SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 bulk lattice constants were calculated and compared with actual experimental data (
Table 2). As shown in
Table 2, the B3LYP calculated ReO
3 bulk lattice constant (3.758 Å) is only overestimated by 0.29% with respect to the experimental value of 3.747 Å [
63]. The, by means of hybrid exchange–correlation functionals, calculated SrZrO
3, BaZrO
3 and PbZrO
3 bulk lattice constants are overestimated with respect to the experimentally measured bulk lattice constants by 0.99%, 0.83% and 1.41%, respectively [
64,
65,
66]. The theoretical ReO
3, SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 [
67] bulk lattice constants were used in all subsequent (001) surface calculations.
The calculated effective charge is +2.382
e for the Re atom in the ReO
3 bulk matrix (
Table 3). The calculated Zr effective charges in the SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 perovskites (+2.174
e, +2.134
e, +2.111
e and +2.144
e, respectively) are similar to each other and strongly different from the Zr formal ionic charge (+4
e). The calculated O effective charge in the ReO
3 bulk is equal to −0.794
e. Ab initio calculated O effective charges in the SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 perovskites are equal to −1.351
e, −1.316
e, −1.160
e and −1.310
e, respectively. Therefore, the SrZrO
3, BaZrO
3 and CaZrO
3 O effective charges are similar, but the O effective charge in the PbZrO
3 crystal is considerably smaller, only −1.160
e (
Table 3). The chemical bond population between Re and O atoms in ReO
3 is equal to +0.212
e. The chemical bond population between Zr and O atoms in SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 matrices are equal to +0.092
e, 0.108
e, 0.106
e, 0.086
e, respectively. Large chemical bond population values between B and O atoms in ReO
3, SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 crystals indicate that the chemical bonding in these materials is covalent.
By means of the B3LYP or B3PW hybrid exchange–correlation functionals, the ReO
3, SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 bulk band gaps at the Γ–Γ point were calculated for the cubic phase of these crystals. It is worth mentioning that the hybrid exchange–correlation functionals, such as B3LYP or B3PW are in excellent agreement with the experimentally obtained band gaps of related ABO
3 perovskites and their (001) surfaces [
5,
16,
47,
48,
68], whereas the density functional theory, consistently underestimates the band gap of complex oxide materials. From another side, it is well known that the Hartree–Fock method considerably overestimates the band gap of complex oxide materials. The B3LYP-calculated bulk band gap for ReO
3 at the Γ-point is equal to 5.76 eV (
Table 4). To the best of our knowledge, there are no reported experimental data for the ReO
3 bulk band gap at the Γ-point. The calculated optical band gap for BaZrO
3 at the Γ-point (4.93 eV) is only underestimated by 6.98% regarding the experimental value of 5.3 eV [
69]. The ab initio calculated optical band gaps at the Γ-point for SrZrO
3, PbZrO
3 and CaZrO
3 perovskite cubic phases are 5.31, 5.63 and 5.40 eV, respectively. Unfortunately, it is not possible to compare the ab initio calculation results for the band gaps at the Γ-point for SrZrO
3, PbZrO
3 and CaZrO
3 perovskites with experimental results, since there are, currently, no reports of the band gaps of SrZrO
3, PbZrO
3 and CaZrO
3 perovskite cubic phases in the literature.
3.2. Ab Initio Calculations of ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) Surfaces
B3LYP or B3PW ab initio calculations for the upper three-layer atom relaxation for the neutral ZrO
2-terminated SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 as well as polar ReO
2-terminated ReO
3 (001) surfaces (
Table 5) were performed. It is worth noting that the ReO
3 material has the cubic ABO
3 perovskite structure and symmetry with the space group number 221, but with the only difference being the A atom vacancy (
Figure 2). For the cases of SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 perovskite ZrO
2-terminated as well as ReO
3 crystal ReO
2-terminated (001) surfaces, according to the ab initio calculations, all upper-layer atoms relax towards the bulk (
Table 5). The ReO
2-terminated ReO
3 (001) surface upper-layer Re atom displacement magnitude (3.19% of
a0) is slightly larger than the ab initio calculated ABO
3 perovskite ZrO
2-terminated (001) surface Zr atom relaxation magnitudes, which are in the range of 1.30% of
a0 for the CaZrO
3 to 2.37% of
a0 for the PbZrO
3 perovskite (
Table 5). In contrast, all SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 perovskite second-layer ZrO
2-terminated (001) surface atoms relax in the outward direction. The only exception to this systematic trend is the second-layer ReO
2-terminated ReO
3 (001) surface O atom inward relaxation towards the bulk; however, this has a small relaxation magnitude, equal to −0.32% of
a0. All the ab initio calculated third-layer atoms for the ZrO
2-terminated SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 as well as ReO
2-terminated ReO
3 (001) surfaces, again, as in the case of the upper-layer atoms, relax inwards, towards the crystal bulk (
Table 5). Nevertheless, the relaxation magnitudes of all first-layer atoms for the ZrO
2-terminated SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 perovskite as well as ReO
2-terminated ReO
3 (001) surfaces are much larger than the relevant relaxation magnitudes of the respective third-layer atoms (
Table 5).
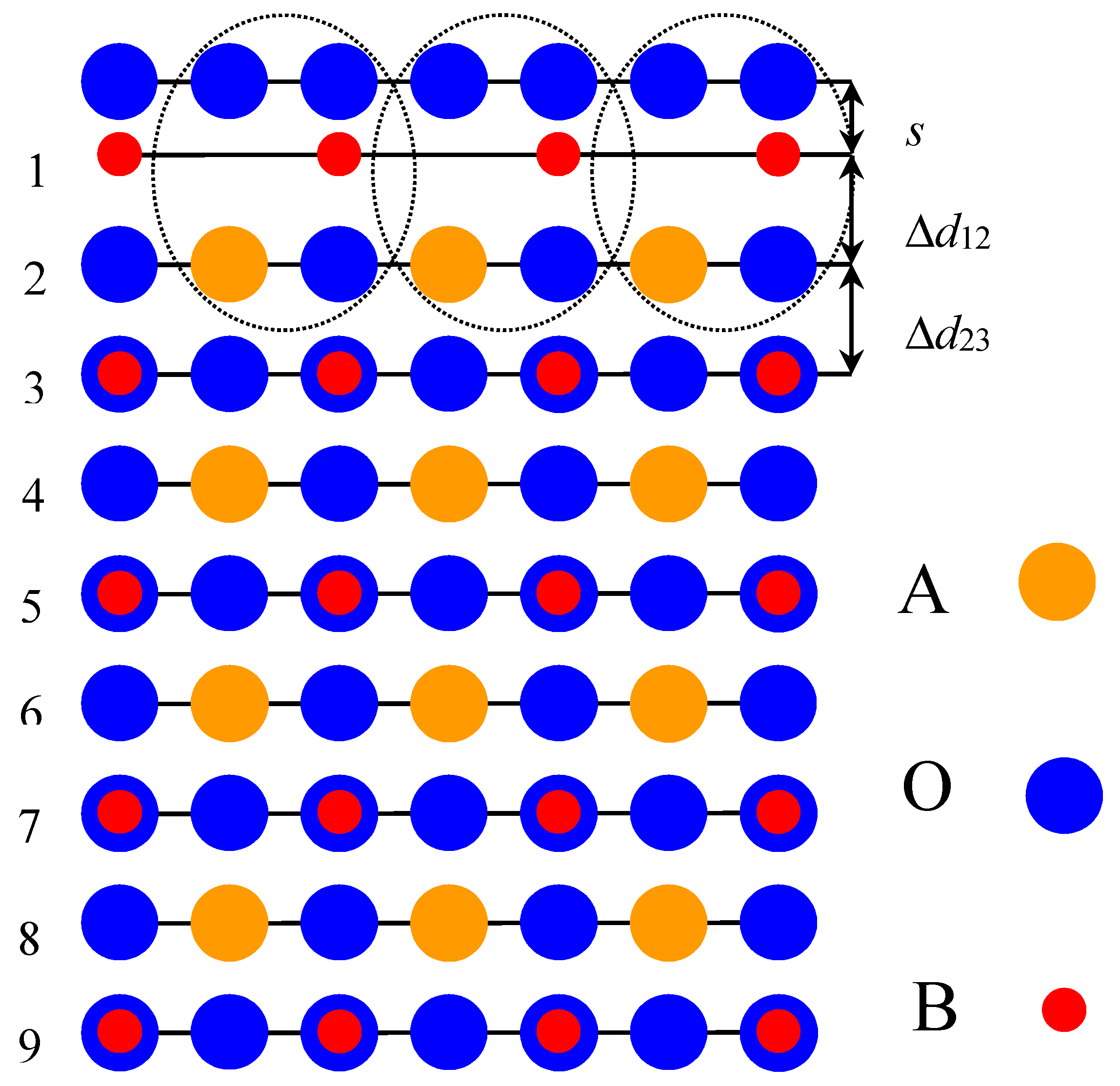
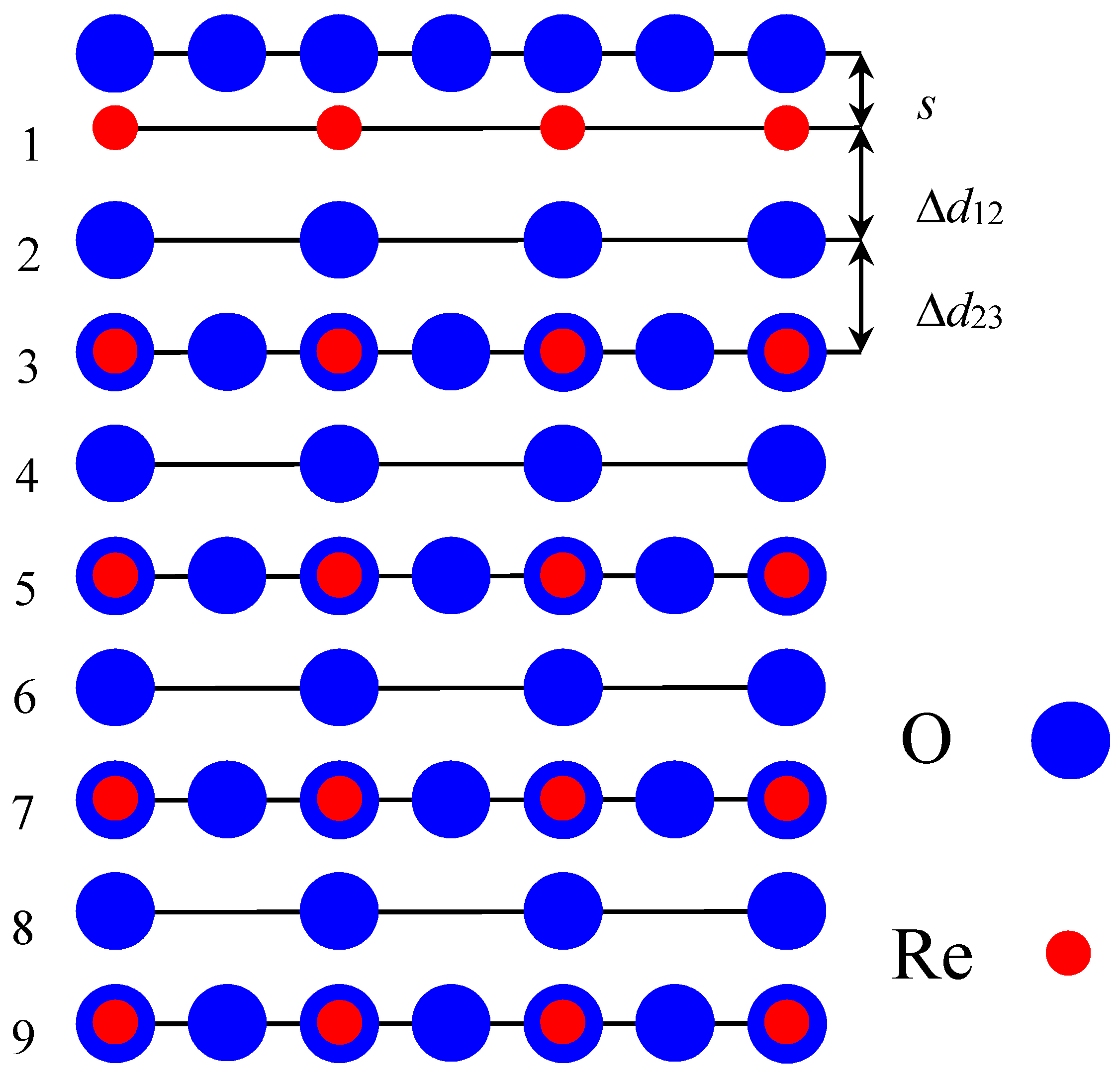
To compare ab initio calculation results for ReO
3, SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 (001) surfaces with the available experimental results, the calculated surface rumplings
s (the relative displacement of oxygen with respect to the metal in the upper surface layer) as well as the changes in interlayer distances, Δ
d12 and Δ
d23, are shown in
Table 6. The calculations of the ReO
3, SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 (001) surface interlayer distances rely on the positions of the metal ions (
Figure 1), which are well known to be much stronger electron scatterers than oxygen ions [
70]. As can be seen from
Table 6, all the calculated ZrO
2-terminated SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 (001) surfaces show the reduction of the interlayer distance Δ
d12 and expansion of Δ
d23. For all ZrO
2-terminated SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 (001) surfaces, the reduction in the interlayer distance, Δ
d12, is larger than the expansion of the respective interlayer distance, Δ
d23. The ab initio calculated surface rumpling,
s, is positive and largest between all calculated surface rumplings (+2.02) for the ReO
2-terminated ReO
3 (001) surface. The calculated surface rumplings,
s, for the ZrO
2-terminated BaZrO
3 and PbZrO
3 (001) surfaces (+0.09 and +0.38, respectively) are also positive, but much smaller than for the ReO
2-terminated ReO
3 (001) surface (+2.02). In contrast, the calculated surface rumplings,
s, for the ZrO
2-terminated SrZrO
3 and CaZrO
3 (001) surfaces are negative (−0.72 and −1.01, respectively).
Unfortunately, to the best of our knowledge, there are no experimental data available for the ReO
3, SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 (001) surface rumpling,
s, as well as interlayer distances, Δ
d12 and Δ
d23. However, such experimental data exist for the related ABO
3 perovskite, SrTiO
3 (
Table 7). To compare the calculated and experimental SrTiO
3 (001) surface structures, the calculated surface rumpling,
s, as well as the changes in interlayer distances, Δ
dij, are detailed in
Table 7. From
Table 7, it can be seen that the agreement is fairly good for all theoretical calculation methods, which all give the same sign for the surface rumpling,
s, as well as the changes in the interlayer distances, Δ
dij. For example, the calculated surface rumpling,
s, for the SrO-terminated surface is much larger than for the TiO
2-terminated SrTiO
3 (001) surface for all theoretical methods [
25,
71,
72,
73,
74,
75]. From
Table 7, it can be seen that both the calculated SrO and TiO
2-terminated SrTiO
3 (001) surfaces always exhibit a reduction in the interlayer distance, Δ
d12 and an expansion of Δ
d23. The theoretically calculated surface rumpling amplitudes,
s, for both SrTiO
3 (001) surface terminations are in fair agreement with the LEED [
70], RHEED [
76], MEIS [
77] and SXRD [
78] experiments (
Table 7). Nevertheless, the calculated changes in interlayer distances disagree with the LEED experiments [
70] for the TiO
2-terminated SrTiO
3 (001) surface, which show an increase in Δ
d12 and reduction in Δ
d23 (
Table 7). In contrast, all ab initio as well as classical shell model calculations show a reduction in the interlayer distance, Δd
12 and an expansion of Δ
d23 (
Table 7). Nevertheless, as can be seen from
Table 7, unfortunately, the different experiments contradict each other with respect to the sign of Δ
d12 and Δ
d23 for the SrO-terminated SrTiO
3 (001) surface and for the sign of Δ
d23 for the TiO
2-terminated SrTiO
3 (001) surface (
Table 7).
The ab initio calculated atomic displacements, the Mulliken static charges as well as bond populations between nearest atoms are reported in
Table 8. The most important effect, as can be seen from
Table 8, is strengthening of the Zr–O chemical bond near the ZrO
2-terminated SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 (001) surface in comparison to the bulk [
79,
80,
81,
82]. In contrast, for the ReO
2-terminated ReO
3 (001) surface, the chemical bond population between the Re and O atoms in the upper surface layer 0.170
e (
Table 8) is slightly smaller than the Re–O chemical bond population in the ReO
3 bulk (0.212
e). Nevertheless, the chemical bond population between the upper-layer Re atom and the second-layer O atom (0.262
e) for the ReO
2-terminated ReO
3 (001) surface is considerably larger than the Re–O chemical bond population in the ReO
3 crystal bulk (0.212
e). It is worth noticing, that the Re and O effective charges in the ReO
3 crystal bulk (+2.382
e for Re and −0.794
e for O) are much smaller than those expected in the ionic model (+6
e for Re and −2
e for O). Moreover, the Re–O chemical bond in the ReO
3 bulk is considerably populated (+0.212
e). It is interesting to note that the Re–O chemical bond population for the ReO
2-terminated ReO
3 (001) surface third layer (+0.208
e) (
Table 8) is already highly similar to the Re–O chemical bond population in the ReO
3 bulk matrix (0.212
e). The Re effective charge in the ReO
2-terminated ReO
3 (001) surface third layer (+2.341
e) is almost as high as the Re effective charge value (+2.382
e) in the ReO
3 bulk crystal. In contrast, the Re effective charge on the ReO
2-terminated ReO
3 (001) surface upper layer, where the surface effect is strong, (+2.258
e) is much smaller than the Re effective charge in the ReO
3 crystal bulk (+2.382
e).
As can be seen from the ab initio calculation results, detailed in
Table 9, the Zr–O chemical bond populations for all four calculated perovskites SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 are larger near their ZrO
2-terminated (001) surfaces than in the bulk. However, the opposite is true for the ReO
3 crystal. The Re–O chemical bond population in the ReO
3 bulk (0.212
e) is larger than it is near the ReO
2-terminated ReO
3 (001) surface (0.170
e). However, it is worth noting that the Re–O chemical bond population between the ReO
2-terminated (001) surface upper-layer Re atom and the second-layer O atom (0.262
e) is considerably larger than the Re–O chemical bond population in the ReO
3 crystal bulk (0.212
e).
The, by means of the hybrid exchange–correlation functionals, calculated bulk band gaps at the Γ–Γ point for ReO
3, SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 crystals are equal to 5.76, 5.31, 4.93, 5.63 and 5.40 eV, respectively (
Table 10). In most cases, there are no experimental data available for the ReO
3, SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 bulk band gaps in the cubic phase. However, the calculated BaZrO
3 band gap at the Γ–Γ point (4.93 eV) is in fair agreement with the experimental data (5.3 eV) [
69]. According to the performed ab initio calculations, the systematic trend is reduction of the ReO
3, SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 bulk band gaps near their ReO
2 or ZrO
2-terminated (001) surfaces, respectively. Namely, the calculated band gap values at the Γ–Γ point for ReO
2-terminated ReO
3 and ZrO
2-terminated SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 terminated (001) surfaces of 0.22, 4.91, 4.48, 4.60 and 5.22 eV, respectively, were always smaller with respect to the bulk band gap value (
Table 10).

 ,
, 



{kind=link}
{kind=link}