1. Introduction
Nature-based small molecular weight ligands, such as stilbenes (1,3-diphenylpropenes) are not only important as anticancer agents, but are also useful for the treatment of various human disorders including diabetes, neurological and cardiovascular diseases, as well as other chronic diseases [
1,
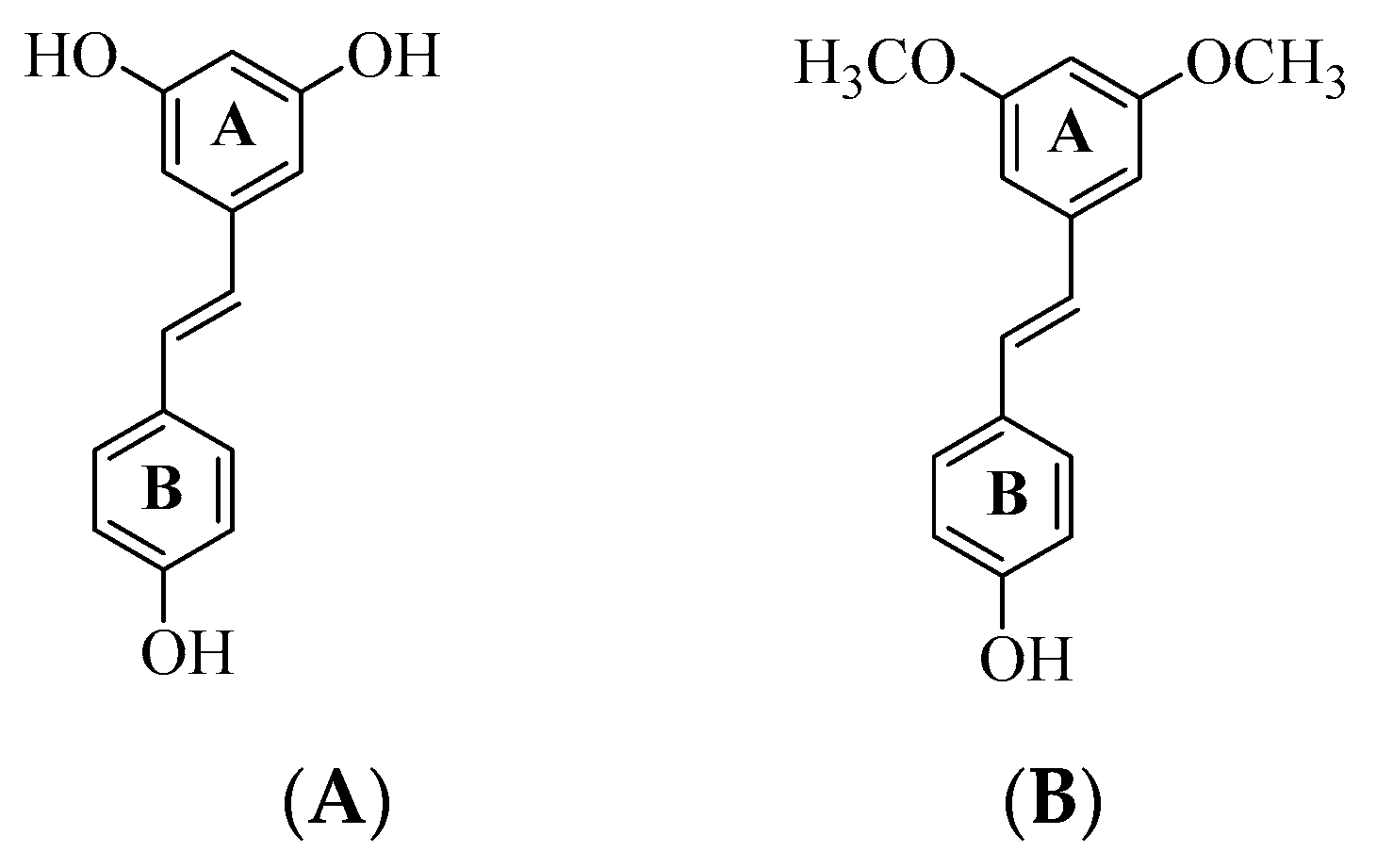
2]. The naturally occurring resveratrol (
trans-3,4,5-trihydroxystilbene) (
A) and pterostilbene (
trans-3,5-dimethoxy-4-hydroxystilbene) (
B) shown in
Figure 1, for example, have been proven to be potent multiple molecular modulators for age-related diseases, including oxidative damage, inflammation, neurodegeneration, obesity, diabetes, and cardiovascular diseases [
2,
3]. Considerable interest has been generated by resveratrol and its analogues due to their antioxidative effects against reactive oxygen species, which are involved in aging and inflammation and cause oxidative damage to biological substances [
2,
3]. Oxidative stress has been linked to neuronal cell death and it is considered to be a fundamental mechanism of progressive neurodegenerative disorders, such as Alzheimer’s disease (AD) [
4]. Despite a wide range of biological activities associated with resveratrol, the application of this stilbene derivative in cosmetics, foods, and drugs has been limited by its poor water solubility, instability, and low bioavailability [
5]. Although generally associated with their traditional use as antibacterial drugs, sulfonamide based compounds have also been found to inhibit cancer related carbonic anhydrase, cysteine protease, HIV protease, cyclooxygenases (COXs), acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) activities [
6]. The sulfonamide group generally adopts tetrahedral geometry, which makes its oxygen atoms to form hydrogen bonds in higher dimensions than amide analogues, for example. Moreover, this group is capable of forming relatively strong electrostatic and hydrogen bonding interactions with protein residues in the receptor binding site and improve the physicochemical properties of the drug molecules [
7]. This moiety in the case of secondary benzenesulfonamide derivatives and sulfonamidochalcones is not only of interest from the medicinal chemistry context [
8,
9,
10,
11,
12], but has been found to provide a versatile template to explore hydrogen bonding interactions [
13,
14,
15,
16,
17,
18] and control molecular conformations [
19,
20,
21,
22,
23].
We considered the molecular construct of resveratrol or pterostilbene and decided to replace the A-ring with an
ortho-(sulfonamido)acetyl group to facilitate intramolecular hydrogen bond formation between the carbonyl oxygen and NH. Integrating this noncovalent interaction in drug design results in conformational restriction of small drug molecules and leads to increased lipophilicity, membrane permeability and pharmacological activity [
24]. One of the limitations associated with the development of more potent drugs for the treatment of neurological disorders is their ability to cross through the blood–brain barrier (BBB), a semipermeable membrane of endothelial cells, which regulates the movement of ions, molecules, and cells between the blood and the brain [
25]. The BBB controls the composition of extracellular fluid in the central nervous system (CNS) and impedes the ability of drugs to cross through to reach target cells and exert their effects [
26,
27,
28]. We envisaged that the replacement of the 4′-hydroxyl group on the B-ring with a lipophilic methoxy or trifluoromethyl group would increase lipophilicity. In our view, initial
N-sulfonylation of 2-amino-5-bromoacetophenone followed by the Suzuki-Miyaura cross-coupling with either 4-methoxyphenylvinylboronic acid or 4-trifluoromethylphenylvinylboronic acid would afford the requisite
N-(2-acetyl-4-(styryl)phenyl)sulfonamide derivatives. These compounds and the corresponding substrates were, in turn, evaluated for inhibitory effect against cholinesterase enzymes (AChE and BChE) and for antioxidant potential. Molecular docking (
in silico) were performed on the most active compound against cholinesterases to determine plausible protein-drug interactions on a molecular level. Their drug likeness (absorption, distribution, metabolism, and excretion: ADMET) properties and ability to cross the blood–brain barrier (BBB) have also been predicted at theoretical level using an in silico method.
2. Materials and Methods
2.1. Materials and Instrumentation
Commercially available reagents were obtained from the vendors and used without further purification. The melting point values of the test compounds are uncorrected, and were recorded on a Thermocouple digital melting point apparatus (Mettler Toledo LLC, Columbus, OH, USA). Gravitational column chromatography was carried out on Merck kieselgel 60 (0.063–0.200 mm) (Merck KGaA, Frankfurt, Germany) as a stationary phase. Nuclear magnetic resonance spectra (
1H-NMR and
13C-NMR spectra) were recorded as deuterated dimethyl sulfoxide ((CD
3)
2SO) solutions using Agilent 500 MHz NMR spectrometer (Agilent Technologies, Oxford, UK) operating at 500 MHz (
1H) and 125 MHz (
13C). The chemical shifts are quoted relative to tetramethylsilane (TMS) used as an internal reference standard (δ = 0.00 ppm) or to residual protonated solvent peak. The high-resolution mass spectra were recorded at an ionization potential of 70 eV using Micromass Autospec-TOF (double focusing high resolution) instrument (Waters Corp., Milford, MA, USA) at the University of Stellenbosch Central Analytical Facility (CAF). The synthesis and analytical data of 2-amino-5-bromoacetophenone (
1) have been reported before [
29].
2.2. Synthesis of N-(2-acetyl-4-bromophenyl)-4-methylbenzenesulfonamide (2)
A solution of 2-amino-5-bromoacetophenone (1.00 g, 4.67 mmol) in pyridine (20 mL) was treated gradually with a 4-methylbenzenesulfonyl chloride (1.2 equiv.). The mixture was stirred under reflux for 2 h followed by quenching with an ice-cold water. The precipitate was filtered and recrystallized from acetonitrile to afford 2 as a white solid (1.51 g, 85%), mp. 162–165 °C; 1H-NMR ((CD3)2SO) 2.31 (3H, s, -C(O)CH3), 2.57 (3H, s, -C6H4CH3), 7.30 (1H, d, J = 8.5 Hz, H-3), 7.34 (2H, d, J = Hz, H-3′,5′), 7.64 (2H, d, J = 8.5 Hz, H-2′,6′), 7.68 (1H, dd, J = 1.5 and 8.5 Hz, H-6), 8.02 (1H, d, J = 1.5 Hz, H-6), 11.06 (1H, s, -NHSO2-); 13C-NMR (D(CD3)2SO) 21.4, 29.2, 116.2, 122.2, 127.4, 130.4, 134.7, 136.0, 137.3137.6, 144.6, 202.4; HRMS (ES): m/z [M + H]+ calc for C15H15BrNO3S: 368.0021; found 368.0010.
2.3. Typical Procedure for the Suzuki-Miyaura Cross-Coupling of 2 to Afford 3a and 3b
A mixture of 2 (0.50 g, 1.36 mmol), PdCl2(PPh3)2 (0.07 g, 0.11 mmol), PCy3 (0.27 g, 0.20 mmol) and K2CO3 (0.27 g, 2.30 mmol) and styrylboronic acid (1.5 equiv.) in 3:1 dioxane–water (v/v; 20 mL) in a two-necked round bottom flask equipped with a condenser and rubber septum was purged with nitrogen gas for 30 min. A balloon filled with nitrogen gas was connected to the top of the condenser, and the reaction mixture was stirred at 70 °C for 3 h. The mixture was quenched with an ice-cold water, and the precipitate was filtered under reduced pressure on a sintered funnel. The residue was purified by column chromatography on silica gel using a 2:1 toluene-ethyl acetate (v/v) mixture as an eluent. Compounds 3a and 3b were prepared in this fashion.
White solid (0.401 g, 70%), mp. 201–203 °C (CH3CN), 1H-NMR ((CD3)2SO) 2.32 (3H, s, -C6H4CH3), 2.63 (3H, s, -C(O)CH3), 3.76 (3H, s, -OCH3), 6.93 (2H, d, J = 8.5 Hz, H-3″,5″), 7.05 (1H, d, J = 16.5 Hz, α-H), 7.20 (1H, J = 16.5 Hz, β-H), 7.34 (2H, d, J = 8.5 Hz, H-3′,5′), 7.38 (1H, d, J = 8.5 Hz, H-3), 7.49 (2H, J = 8.5 Hz, H-2′,6′), 7.66 (2H, J = 8.5 Hz, H-2″,6″), 7.74 (1H, ddd, J = 1.5 and 8.5 Hz, H-4), 8.06 (1H, d, J = 1.5 Hz, H-6), 11.20 (1H, s, -NHSO2-); 13C-NMR ((CD3)2SO) 21.4, 29.1, 55.6, 114.7, 120.1, 124.7, 125.1, 127.4, 128.3, 129.1, 129.9, 130.4, 130.6, 131.9, 133.5, 136.2, 137.3, 144.9, 159.6, 203.8; HRMS (ES): m/z [M + H]+ calc for C24H24NO4S: 422.1424; found 422.1420.
(E)-N-(2-Acetyl-4-(4-(trifluoromethyl)styryl)phenyl)-4-methylbenzenesulfonamide (3b)
White solid (0.525 g, 85%), mp. 188–190 °C (CH3CN), 1H-NMR ((CD3)2SO) 2.32 (3H, s, -C(O)CH3), 2.65 (3H, s, -C6H4CH3), 7.35 ((1H, d, J = 16.5 Hz, H-β), 7.36 (2H, d, J = 8.5 Hz, H-3″,5″), 7.40 (1H, d, J = 16.5 Hz, H-α), 7.43 (1H, d, J = 8.5 Hz, H-3), 7.70 (2H, d, J = (2H, d, J = 8.5 Hz, H-2″,6″), 7.76 (2H, d, J = 8.5 Hz, H-3′,5′), 7.82 (2H, d, J = 8.5 Hz, H-2′,6′), 7.82 (1H, dd, J = 1.5 and 8.5 Hz, H-4), 8.17 (1H, d, J = 1.5 Hz, H-6), 11.30 (1H, s, -NHSO2-); 13C-NMR ((CD3)2SO) 21.4, 29.1, 119.8, 124.2 (q, JCF = 270.5 Hz), 124.7, 126.1 (q, 3JCF = 3.8 Hz), 127.3, 127.4, 128.0, 128.2, 130.0, 130.4, 131.5, 132.3, 132.6, 136.2, 138.2, 141.4, 144.6, 203.7; HRMS (ES): m/z [M + H]+ calc for C24H21F3NO3S: 460.1232; found 460.1103.
2.4. Data Collection and Refinement
Intensity data was determined on a Bruker Venture D8 Photon CMOS diffractometer with graphite-monochromated MoKa
1 (l = 0.71073 Å) radiation at 173 K using an Oxford Cryostream 600 cooler. Data reduction was carried out using the program SAINT+, version 6.02 [
30] and empirical absorption corrections were made using SADABS [
30]. Space group assignments was made using
XPREP [
30]. The structure was solved in the
WinGX [
31] Suite of programs using intrinsic phasing through
SHELXT [
32] and refined using full-matrix least-squares/difference Fourier techniques on
F2 using
SHELXL-2017 [
33]. All C bound hydrogen atoms were placed at idealized positions and refined as riding atoms with isotropic parameters 1.2 times or 1.5 times those of their parent atoms. N-bound hydrogen atoms were located in the difference Fourier map and their coordinates and isotropic displacement parameters refined freely. Diagrams and publication material were generated using
Olex2 [
31], and
Mercury [
34]. Crystal data and structure refinement for compounds
2,
3a and
3b are included as
Table S1 in the Supplementary Information (SI).
2.5. Hirshfeld Surface Analyses
The Hirshfeld surface analyses were obtained using Crystal Explorer software [
35]. The normalized contact distance (
dnorm) is based on the distance from the point to the nearest nucleus external to the surface (
de) and the distance to the nearest nucleus internal to the surface (
di) [
36].
2.6. Cholinesterase Inhibition Assays of 1, 2, 3a and 3b
AChE and BChE as well as the corresponding substrates were obtained through BIOCOM Africa (Pty) Ltd. (Centurion, Pretoria, South Africa). Cholinesterase activities were determined following a modification of the Ellman’s method as described in our previous study [
37], and the reactions were performed in triplicate at 37 °C in a 96-well plate. The stock solutions (200 µM) of both the test compounds and reference standard (donepezil) were prepared in DMSO, and further diluted to 5, 10, 25, 50 and 100 µM using Tris buffer (50 mM; pH 7.7).
2.6.1. AChE Inhibition Assay of 1, 2, 3a and 3b
Reactions were performed in a 96-well plate where the following was added sequentially, 8.0 µL solution of the test compound, 2.0 µL of AChE (0.04 mg/mL), and 70 µL of Tris buffer (C4H11NO3, 50 mM, pH 7.7). The enzyme and inhibitor mixture were preincubated for 30 min at room temperature followed by addition of 10 µL of 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB, 3 mM in Tris buffer, 50 mM, pH 7.7) and 10 µL of acetylcholine iodide (AChI, 5 mM in Tris buffer, 50 mM, pH 7.7) to each well to initiate the reaction. Five different absorbance readings (412 nm) were recorded for each of three replicates of a single reaction using a Varioskan flash spectrophotometer plate reader (Thermo Scientific, Waltham, MA, USA). The average values obtained from the absorbance readings were used to determine the IC50 and standard deviation values using the Graph Pad Prism.
2.6.2. BChE inhibition assay of 1, 2, 3a and 3b
In a 96-well plate 8.0 µL of the test compound and 2.0 µL of BChE (0.02 mg/mL), were incubated at room temperature for 30 min. Tris buffer (70 µL) was added to each well containing the reaction mixture and incubation was continued for further 10 min at this temperature followed by addition of 10 µL solution of 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) (3 mM in Tris buffer, 50 mM, pH 7.7) and butyrylcholine iodide (5 mM in Tris buffer, 50 mM, pH 7.7) to each well to initiate the reaction. Five different absorbance readings (412 nm) were recorded for each of three replicates of a single reaction using a Varioskan flash spectrophotometer plate reader, and the IC50 and standard deviation values were obtained using the Graph Pad Prism.
2.7. Antioxidant Assay
2.7.1. Determination of Reducing Activity of the Stable DPPH Radical by 1, 2, 3a and 3b
The antioxidant activities of the test compounds against ascorbic acid (Sigma Aldrich, Saint Louis, MI, USA) as a positive control were evaluated using 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging assay developed by Zhu et al. as described in our previous study [
37]. The experiment was done in triplicate with ascorbic acid (vitamin C) used as a positive control for the assay. Solutions (20 µL) of the test compounds and ascorbic acid in DMSO (final concentrations: 5, 10, 25, 50 and 100 µM) were added into each designated well of a 96-well plate. A solution of 0.20 mM DPPH (20 µL) in methanol was added to each well, and the 96-well plate was wrapped with aluminium foil and incubated in the dark for 45 min. Five absorbance readings were recorded at 512 nm using Varioskan flash microplate spectrophotometer reader. The average values obtained from the absorbance readings were used to determine the IC
50 and standard deviation values.
2.7.2. NO Free Radical Scavenging Assay
Nitric oxide was generated from sodium nitroprusside and measured by Griess’ reaction following the literature method [
38]. The experiment was done in triplicate with ascorbic acid (vitamin C) used as a positive control for the assay. In a 96 well plate 50 μL of the test compounds (0.1, 0.5, 1.0, 5.0 and 10.0 μM in methanol) or the positive control were mixed with 50 μL of sodium nitroprusside (10 mM) prepared in phosphate buffered saline (pH = 7.4) and allowed to incubate at room temperature for 3 h. Afterwards, 300 μL of the Griess reagent (a mixture of 0.2%
N-(1-naphthyl)ethylenediamine dihydrochloride and 2% sulfanilamide in 5%) was added to each well and the absorbance recorded at 546 nm using Varioskan flash microplate spectrophotometer reader.
2.8. Computational Studies
2.8.1. Molecular Docking of Test Compounds into AChE and BChE
The PDB structure codes 4EY7 and 1P0I representing AChE and BChE were prepared using the prepare protein protocol with default settings prior to docking using Discovery Studio software version v20.1.0.19295 (Accelrys, San Diego, USA). The binding site x, y and z coordinates used for docking were −14.0, −44.0, 27.9 with a radius of 10 for 4EY7 which correspond to the site where donepezil is bound in the pdb structure. The binding site identified as a receptor cavity site 1 was used for 1P0I with x, y and z coordinates 134, 115, 40.5 and radius of 12. Compounds 1, 2, 3a and 3b were drawn and prepared using the prepare ligand protocol with default settings. Donepezil was also drawn and prepared in the same way as the compounds and docked as a reference standard. Docking was performed using the CDOCKER module and the top poses with the optimal CDOCKER and CDOCKER interaction energy, with no unfavourable interactions were selected. The selected pose in each case was then optimised by conducting a binding energy calculation with default settings except that the amino acid residues in the binding site were allowed to flex and Generalized Born with Molecular Volume (GBMV) was used as an implicant solvent model.
2.8.2. Physicochemical Parameters of 1, 2, 3a and 3b
The bioactivity score and the molecular properties of the test compounds were calculated by Molinspiration (
www.molinspiration.com). The Lipinski’s rule of five was used to evaluate the druglikeliness of the test compounds.
3. Results and Discussion
Since
p-toluenesulfonamide has previously been found to exhibit anticancer properties with good lipophilicity [
39], we subjected the known 2-amino-5-bromoacetophenone (
1) to sulfonylation with
p-toluenesulfonyl chloride in pyridine under reflux for 2 h to afford
N-(2-acetyl-4-bromophenyl)-4-methylbenzenesulfonamide (
2), exclusively (
Scheme 1). High temperature and prolonged time in the case of the reaction of the analogous 2-aminobenzamide (anthranilamide) with alkyl/arylsulfonyl chlorides, on the other hand, have previously been found to facilitate the attachment of a second sulfonyl group on the aniline nitrogen atom and/or dehydration of the amide NH
2 group into a nitrile group [
40,
41]. The
1H NMR spectrum of
2 showed the presence of an intense singlet at δ = 2.31 ppm integrating for three protons, which corresponds to the methyl group and a set of intense doublets in the aromatic region each integrating for two protons for the
para substituted phenyl ring of the incorporated
p-toluenesulfonyl group. The singlet for the sulfonyl proton (NH) resonates significantly downfield at δ = 11.06 ppm due to deshielding effect of the S=O bonds, which is also consistent with the presence of intramolecular hydrogen bond. The presence of this singlet ruled out the possibility of the attachment of a second sulfonyl group on the aniline nitrogen atom. The compound
2 was, in turn, subjected to the Suzuki-Miyaura cross-coupling with 4-methoxyphenylvinylboronic acid or 4-trifluoromethylphenylvinylboronic acid in the presence of dichlorobis(triphenylhosphine)palladium(II) (PdCl
2(PPh
3)
2)) as source of active Pd(0) species, tricyclohexylphosphine (PCy
3) as a ligand and cesium carbonate as a base in aqueous dioxane under reflux for 3 h to afford
3a and
3b, respectively. The
1H NMR and
13C NMR spectra of
3a and
3b (copies of spectra are included as
Figure S1 in the Supplementary Information, SI) showed the presence of increased number of signals in the aromatic region compared to the corresponding spectra of
2. The olefinic protons of the styryl wing of compounds
3 resonate as two sets of doublets among the aromatic proton signals with vicinal coupling constant (
J) values of 16.5 Hz consistent with the
trans geometry about the ethylene linkage. Although the presence of the double bond in stilbene derivatives may give rise to
trans- and
cis-isomeric forms, the
trans geometry is the thermodynamically more stable and favourable geometry [
2]. A singlet for NH of compounds
3 resonates significantly downfield (around δ = 12.04 ppm) compared to that of
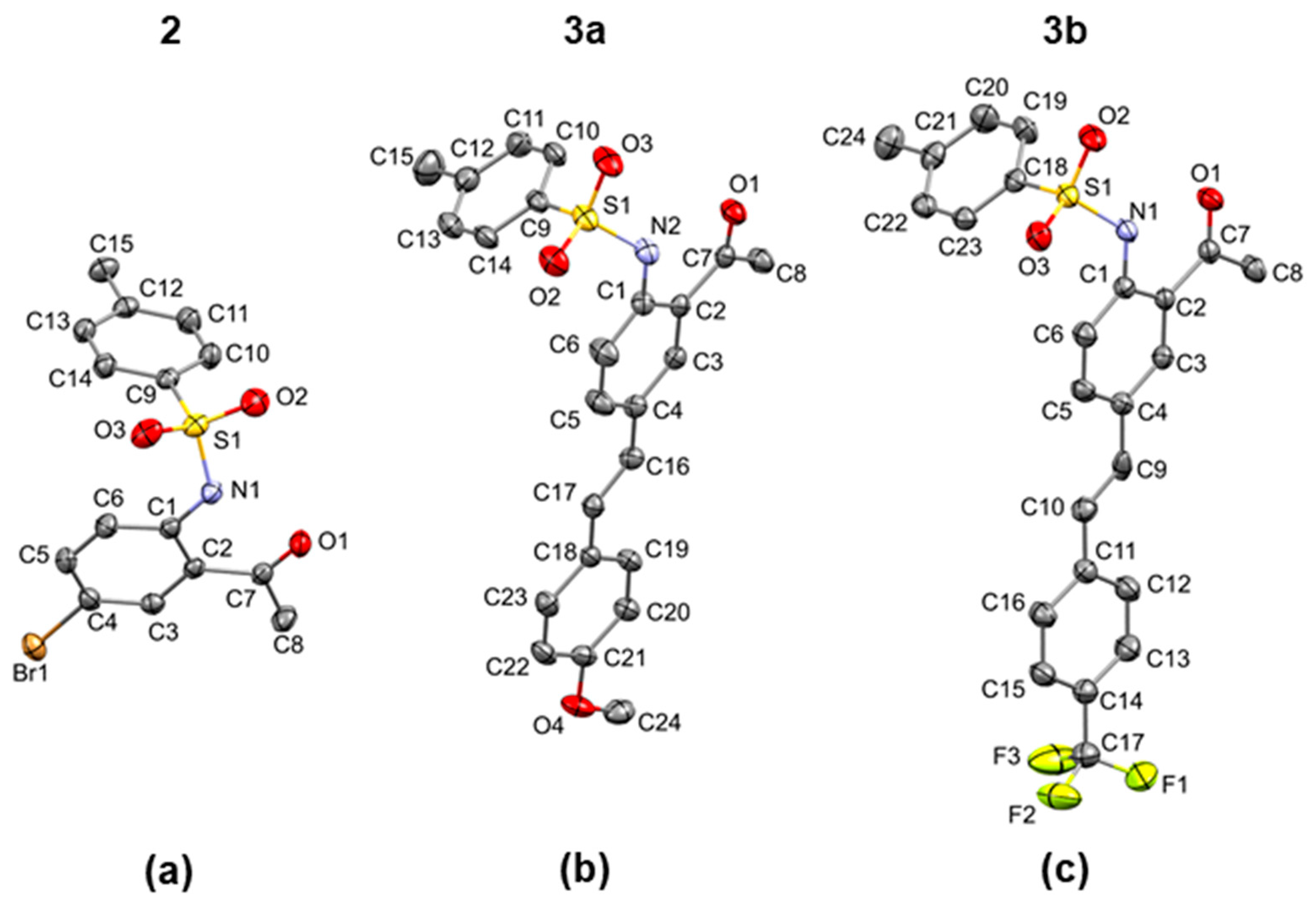
2 due to conjugative effect of the styryl wing. After confirming these compounds homogeneity and purity by NMR and high-resolution mass spectroscopic techniques, the compounds were recrystallized from acetonitrile to obtain single crystals suitable for X-ray diffraction (XRD) analysis. XRD method confirmed the presence of thermodynamically more favourable six-membered intramolecular hydrogen bonding motif designated as S(6) in the graph-set assignments [
42] involving interaction between NH of the sulfonamide group and the carbonyl oxygen of
2 (
Figure 2a),
3a (
Figure 2b) and
3b (
Figure 2c). A list of the corresponding N–H
...O bond distance and angle values, and other significant geometric parameters (bond distances and angles) is shown in
Table 1. The hydrogen bond distance values of 1.93(2) Å (
2), 1.90(2) Å (
3a) and 1.98(3) Å (
3b), which are lower than 2.00 Å may be considered as candidates to stronger hydrogen bonds. The presence of a thermodynamically more favourable six-membered intramolecular hydrogen bonding motif between the carbonyl oxygen and N-H has also been observed before for the analogous
ortho-(4-tolylsulfonamido)benzamides [
43] and the
ortho-(benzenesulfonamido)chalcone derivatives [
8,
10,
14,
16]. XRD analysis also confirmed the
trans geometry of the styryl arm of these compounds. The distorted tetrahedral geometry of sulfur atom resulted in a twisted conformation of the sulfonamido group of these compounds from co-planarity of the acetophenone (
2) or 5-styrylacetophenone (
3a and
3b) scaffolds. The torsion angle C(1)-N(2)-S(1)-C(9) between the two rings of compound
2 is -53.97(13)°, while that between the planar styrylacetophenone framework and the 4-tolyl ring of
3a and
3b are C(1)-N(2)-S(1)-C(9) of -52.51(15)° and C(1)-N(1)-S(1)-C(18) of -61.30(2)°, respectively. The conformations of the N(2)–C(1) bond in the C–SO
2–NH–C segment of the structure of compounds
2 and
3 are
trans and
gauche with respect to the S(1)=O(2) and S(1)=O(3) bonds, respectively. The deviation of the sulfonamide moiety from co-planarity of a conjugated framework is a common phenomenon observed in the crystal structures of the benzene sulfonamide derivatives [
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
41,
43]. This enables the sulfonamide group to make dimer and catemer motifs of N-H
…O hydrogen bonds.
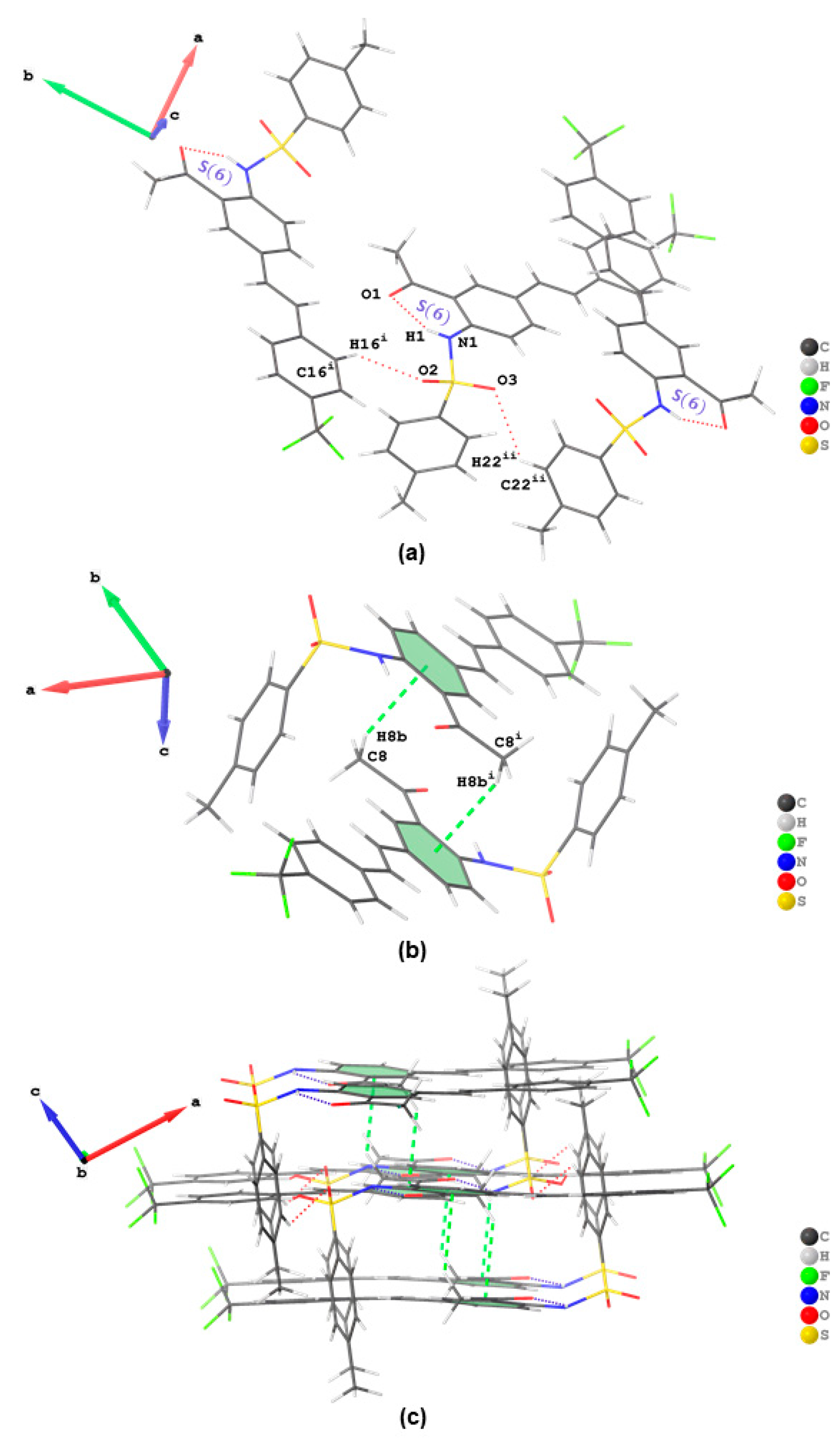
The geometry of compounds
1–
3 is interesting from medicinal chemistry point of view because six-membered intramolecular hydrogen bonded drug-like molecules have been found to exhibit favourable alignment with the protein pocket to lead to increased ligand-receptor interactions [
44]. Non-covalent interactions, such as hydrogen bonding, halogen bonding and aromatic-aromatic (π···π, CH···π stacking) interactions as well as other weak contacts play crucial roles in various fields of medicinal chemistry and materials science [
45,
46]. The study of these cooperative and competing non-covalent interactions is crucial to extrapolate structure activity relationships (SAR) and these play a significant role in layered materials [
47]. The intermolecular hydrogen bonding patterns of aromatic sulfonamides are classified into four types, namely, dimeric, zigzag, helical, and straight patterns, all of which retain the synclinal conformation of the sulfonamide functionality [
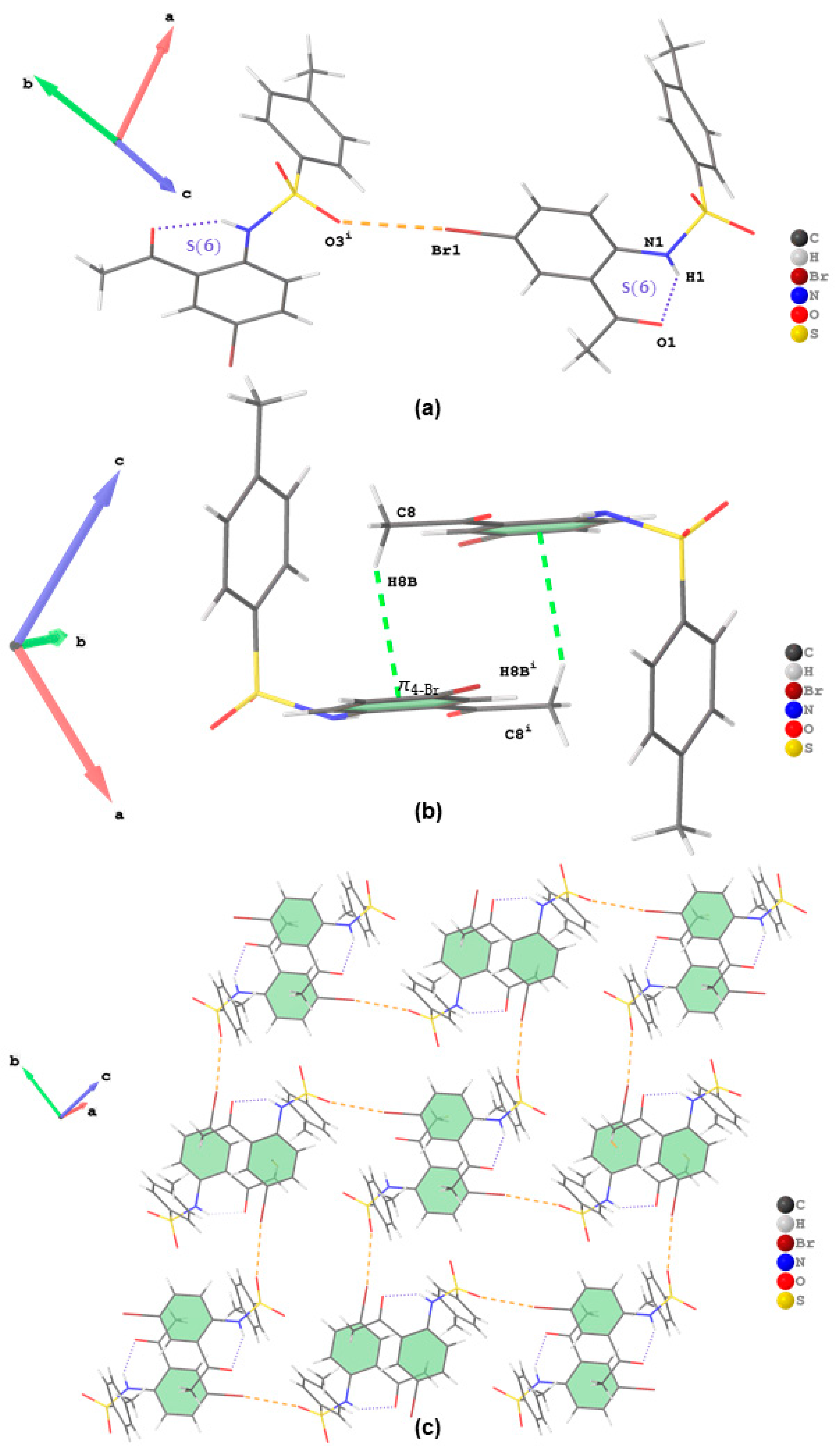
21]. The representation of intermolecular interactions and packing diagrams of
2,
3a and
3b are presented in
Figure 3,
Figure 4 and
Figure 5, respectively. Their packing diagrams reveal the presence of intramolecular N—H···O hydrogen bonds with
graphset descriptors characteristic of the
ortho-aminoacetyl derivatives. Intermolecular Br···O halogen bonds (Br···O = 2.967(2) Å; symmetry code: -1/2+
x,3/2-
y,-1/2+
z) exist in the crystal packing of
2 between bromine atom and one of the oxygen atoms of the sulfonamide moiety (
Figure 3a). The twisting of the sufonamide moiety facilitated parallel alignment of the acetophenone scaffold of adjacent molecules to engage in weak intermolecular C—H···π interactions involving the hydrogen atom of the acetyl group of one molecule and the acetophenone ring of the neighbouring partner (
Figure 3b). A combination of intramolecular hydrogen bonding, halogen bonding and C—H···π interactions resulted in the formation of 2D supramolecular structures that extend diagonally with respect to the crystallographic
ac plane in the crystal packing of
2 (
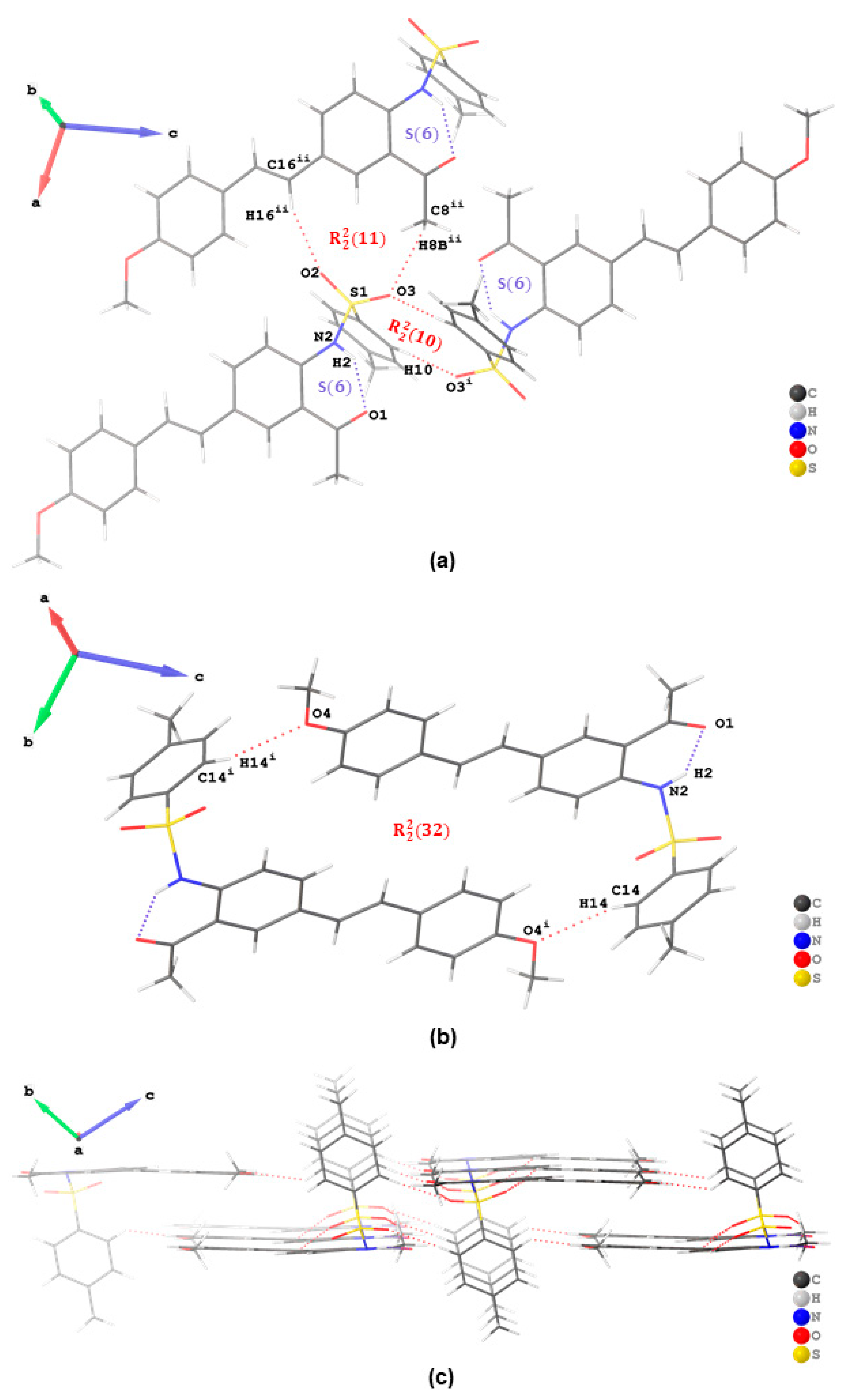
Figure 3c). In addition to a six-membered ring formed from the classical intramolecular hydrogen bond, the crystal packing of
3a is further stabilized by intermolecular hydrogen bonds of the type C—H···O with
and
graphset descriptors (
Figure 4a). There is additional stabilization of this network by weak hydrogen bonding interaction between a hydrogen atom
ortho to -SO
2- and oxygen atom of the methoxy group of another molecule, i.e., C(14)–H(14)···O(4) interaction, in the dimeric assembly resulting in an ring motif with
graphset descriptor (
Figure 3b). A 2D supramolecular network is stabilized by a combination of intramolecular and intermolecular hydrogen bonding interactions without aromatic-aromatic interaction (
Figure 4c). The crystal packing of
3b is also stabilized by intramolecular hydrogen bond and by a combination of intermolecular interactions of the type C—H···O (
Figure 5a) and C—H···π interactions (
Figure 5b) resulting in the formation of 2D supramolecular structures in its crystal packing (
Figure 5c). Similar interactions were also found to be predominant in the Hirshfeld surfaces of the analogous
ortho-sulfonamido chalcones [
8,
10,
14,
16]. Intermolecular C—H···Br and C—H···F hydrogen bonding patterns were also found in the halogen-containing compounds
2 and
3b, respectively (refer to
Figure S2 in SI).
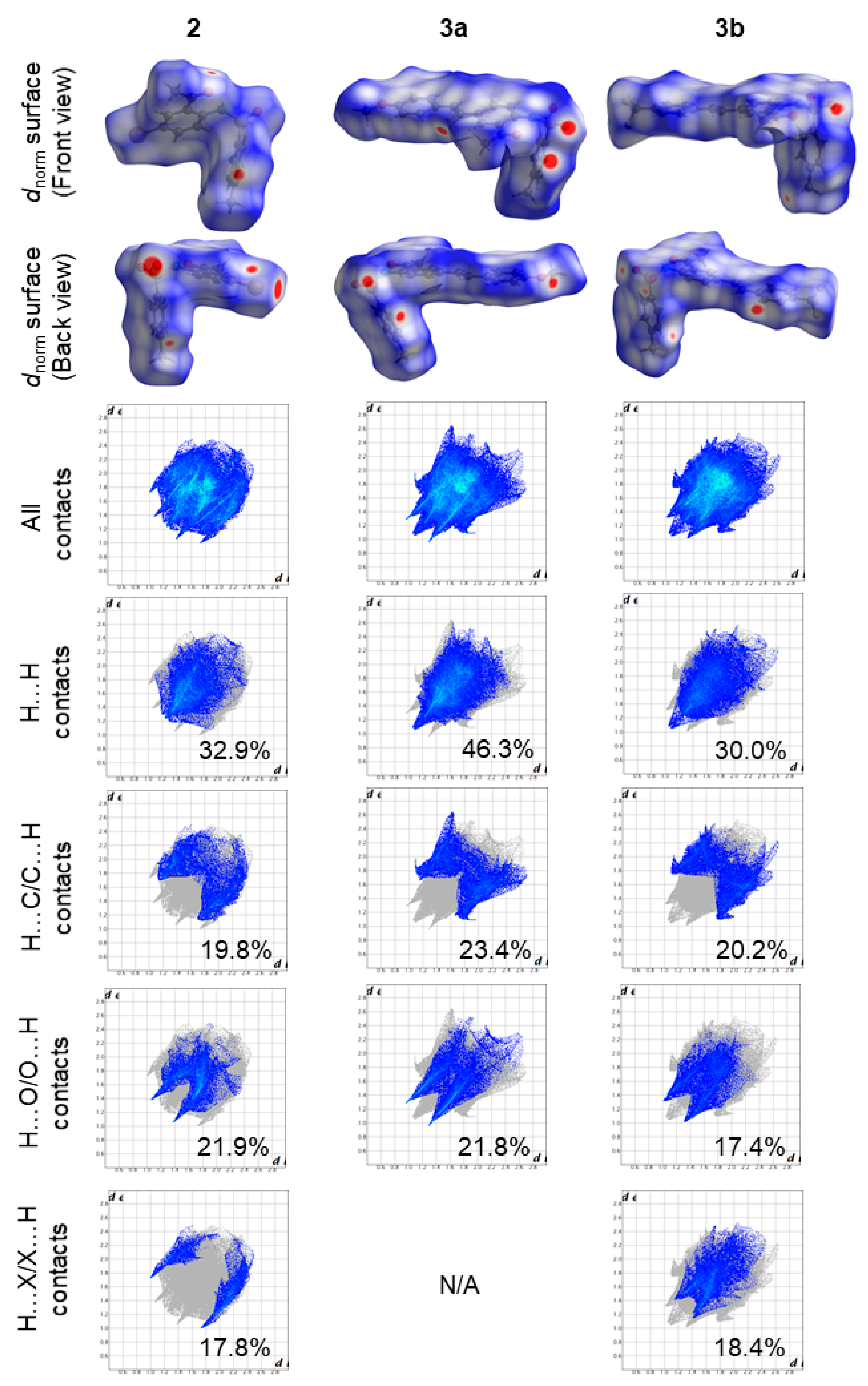
Hirshfeld topology analyses of compounds
2,
3a and
3b were performed to quantify the intermolecular interactions between molecules in the crystal packing. The Hirshfeld
dnorm surface maps and the corresponding fingerprint plots for the H···H, H···O/O···H, C···H/H···C, and H···X/X···H (where X is the corresponding halogen in
2 and
3b) interactions in all compounds, are depicted in
Figure 6. In all these compounds, the H···H contacts contribute significantly towards the Hirshfeld surfaces (30–46.3%) compared to the rest of the contacts. Compound
3a had the highest contribution of H···H contacts due to the presence of the electron donating
para-methoxy substituent on the styryl moiety, which increases the hydrogen composition. Interestingly, it appears that the presence of a halogen atom in
2 and
3b significantly decreases the contribution of H···H contacts as the H···X/X···H increases due to the occurrence of intermolecular C—H···Br (in
2) and C—H···F (in
3b) hydrogen bonds. This is more evident when comparing
3a and
3b whereby a 16.3% difference in the contribution of H···H contacts and a rise of 18.4% in H···X/X···H contacts was observed. Consequently, the high hydrogen composition in
3a also resulted in the highest reciprocal H···C contacts contribution (23.4%) towards the Hirshfeld surface in this work. The reciprocal H···C contacts were attributed to intermolecular C—H···π interactions observed in the crystal packing of each compound. The red areas on the Hirshfeld surface around the oxygen atoms are due H···O/O···H contacts and are attributed to intermolecular C—H···O hydrogen bonds. The contribution of the reciprocal H···O contacts is similar in
2 (21.9%) and
3a (21.8%). However, the reciprocal H···O contacts in
3b (17.4%) is significantly lower than that in
2 and
3a, and this could be due to the presence of the
para-trifluoromethyl substituent on the styryl moiety of
3b. Other prominent red areas on the Hirshfeld surface of
2 were observed around the bromine and oxygen atoms. These red areas correspond to Br···O contacts due to the presence of intermolecular halogen (Br···O) bonds and contribute the least towards the Hirshfled surface in
2 (4.8%). The crystal packing of
3b, on the other hand, is further stabilized by nonclassical hydrogen (C—H⋯F) bonds with no halogen bonding detected or observed.
Sulfonamide derivatives have previously shown very good inhibition against cholinesterase (AChE and BChE) enzymes [
48]. Our interest in the development of compounds with anti-Alzheimer’s properties encouraged us to evaluate compounds
1–
3 through enzymatic assays
in vitro for inhibitory effect against AChE and BChE, and for antioxidant potential in the DPPH and NO radical scavenging assays. Donepezil and ascorbic acid were used as reference standards for the enzyme and antioxidant assays, respectively. The corresponding IC
50 values of three independent experiments are represented in
Table 2. 2-Amino-5-bromoacetophenone
1 was found to exhibit reduced inhibitory effect against AChE and BChE activities compared to donepezil (IC
50 = 1.24 ± 0.15 µM and 3.12 ± 0.18 µM, respectively) with IC
50 values of 12.6 ± 0.20 µM and 14.6 ± 0.32 µM, respectively. The presence of a sulfonamide moiety in
2 resulted in improved activity against AChE (IC
50 = 8.9 ± 0. 21 µM) and significantly reduced effect against BChE (IC
50 = 26.5 ± 0.24 µM) compared to the parent compound
1. However, the sulfonamide moiety resulted in reduced free radical scavenging activity of
2 in the DPPH and NO assays compared to
1 (IC
50 = 12.3 ± 0.21 µM and 7.4 ± 0.16 µM, respectively) with IC
50 values of 20.6 ± 0.42 µM and 15.7 ± 0.20 µM, respectively. The corresponding IC
50 values for ascorbic acid are 4.65 ± 0.13 µM and 6.23± 0.13 µM, respectively. When a styryl group was introduced on the scaffold of
2, the corresponding derivatives
3a and
3b were found to strongly impair the activity of both enzymes compared to their precursors. Replacement of bromine atom with a lipophilic 4-methoxystyryl group in
3a resulted in significantly improved inhibitory effect against AChE and BChE with IC
50 values of 4.3 ± 0.23 µM and 5.6 ± 0.24 µM, respectively. Although less active than
3a, the 5-(4-trifluorostyryl) substituted derivative
3b also exhibited significant activity against AChE (IC
50 = 6.2 ± 0.21 µM) and moderate effect against BChE (IC
50 = 10.5 ± 0.47 µM) compared to its precursors. Both styryl derivatives
3a and
3b exhibited moderate and significant free radical scavenging properties in the DPPH (IC
50 = 16.5 ± 0.31 and 13.9 ± 0.10 µM, respectively) and NO assays (IC
50 = 9.6 ± 0.45 and 11.9 ± 0.31 µM, respectively). The presence of styryl and lipophilic sulfonamide groups in
3a and
3b resulted in dual anticholinesterase effect and antioxidant activities consistent with the design strategy.
IC50 values (μM) were calculated from log dose inhibition curves and are expressed as means ± standard deviation (SD) of three independent experiments.
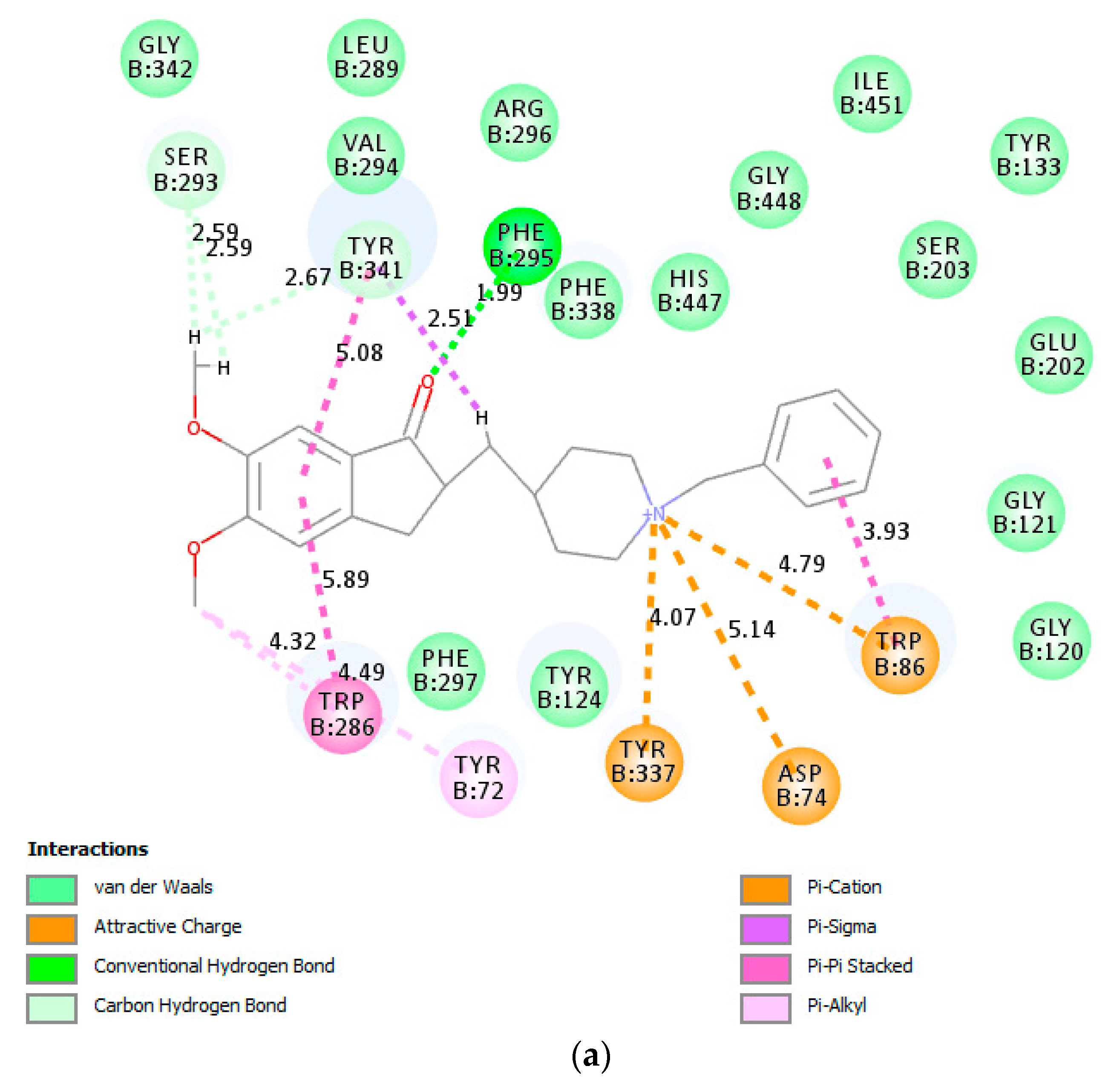
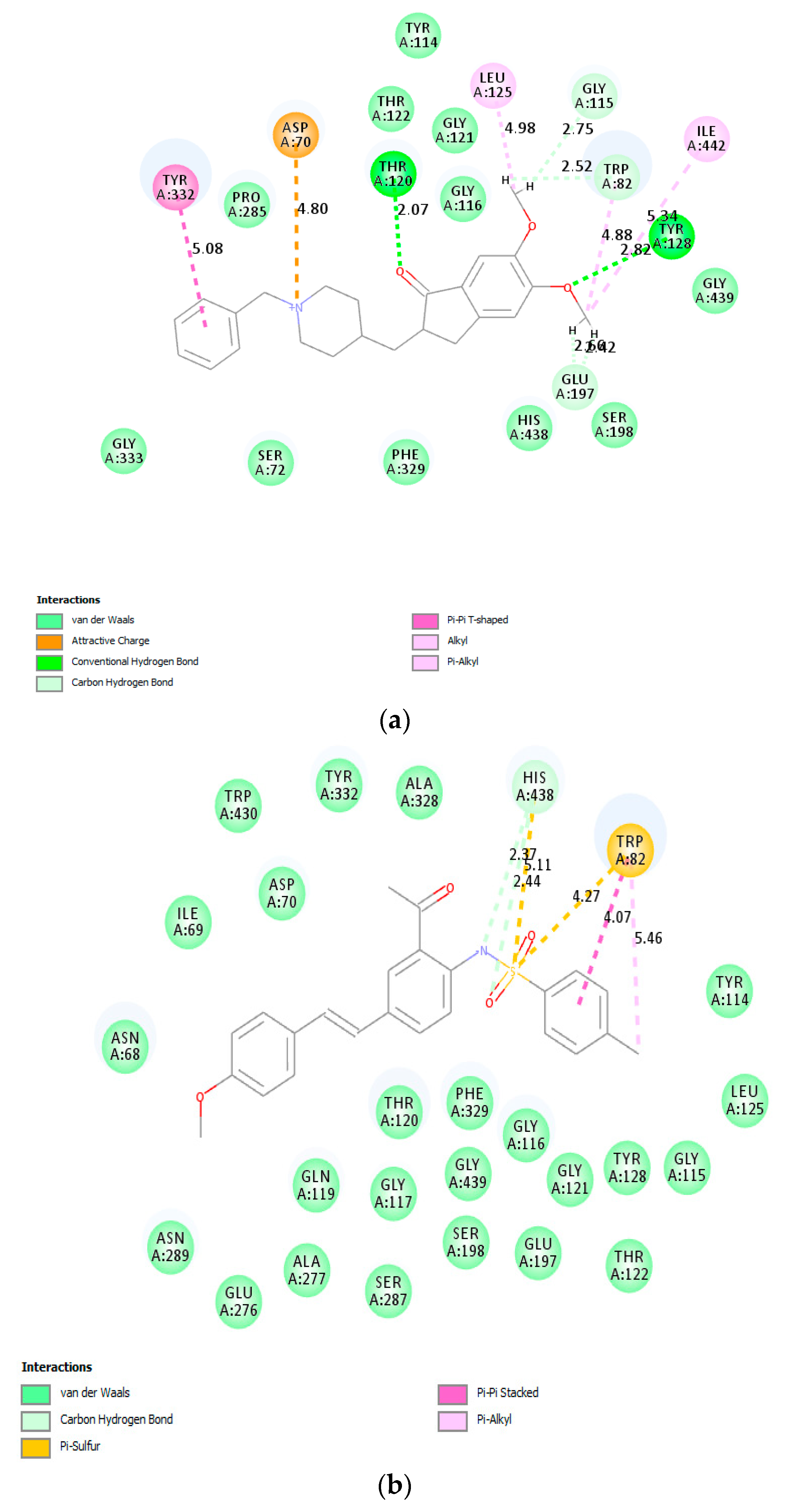
In an attempt to figure out the plausible protein–ligand interactions at molecular level, we performed molecular docking studies on donepezil and
3a against both enzymes. The top scoring docked pose of donepezil into AChE crystal (PDB code: 4EY7) was applied as starting point for molecular docking.
Figure 7a shows donepezil interacting with both the catalytic anionic site (CAS) featuring Trp86 residue and the peripheral anionic site (PAS) including Asp74, Trp286 and Tyr341 residues. The indanone moiety of donepezil is involved in π···π, and π···alkyl interaction with Trp286 residue in the peripheral anionic site (PAS), while the benzyl ring features π···π and π···cation interaction with Trp86 residue in the catalytic anionic site (CAS). Moreover, the carbonyl group of the indanone moiety is involved in hydrogen bonding with Phe295 residue. The compound
3a was docked into the active site of AChE using the same parameters and site as for the docking of donepezil. The docking pose of
3a shows no conventional hydrogen bonding interaction with the protein residues of AChE (
Figure 7b). The carbonyl oxygen is, however, involved in weak carbon hydrogen bonding interactions with Gly121 and Gly122. The benzamide ring is involved in π···π stacking interactions with Tyr337 and T-shaped π···π stacking interaction with Tyr341. The
p-tolyl ring of the styryl arm of this compound is also involved in π···π stacking interaction with Trp286 and Tyr341. The latter residue and Ser293 are involved in weak van der Waals and carbon hydrogen bonding interactions with the hydrogen atoms of methoxy group. Sulfur atom of this compound is involved in π···sulfur interaction with the CAS residues Trp86 and His447. His447 is also involved attractive charge interaction with nitrogen atom of the sulfonamide moiety. The binding of
3a with AChE was mainly provided due to the presence of hydrophobic interactions with no conventional hydrogen bonding interactions.
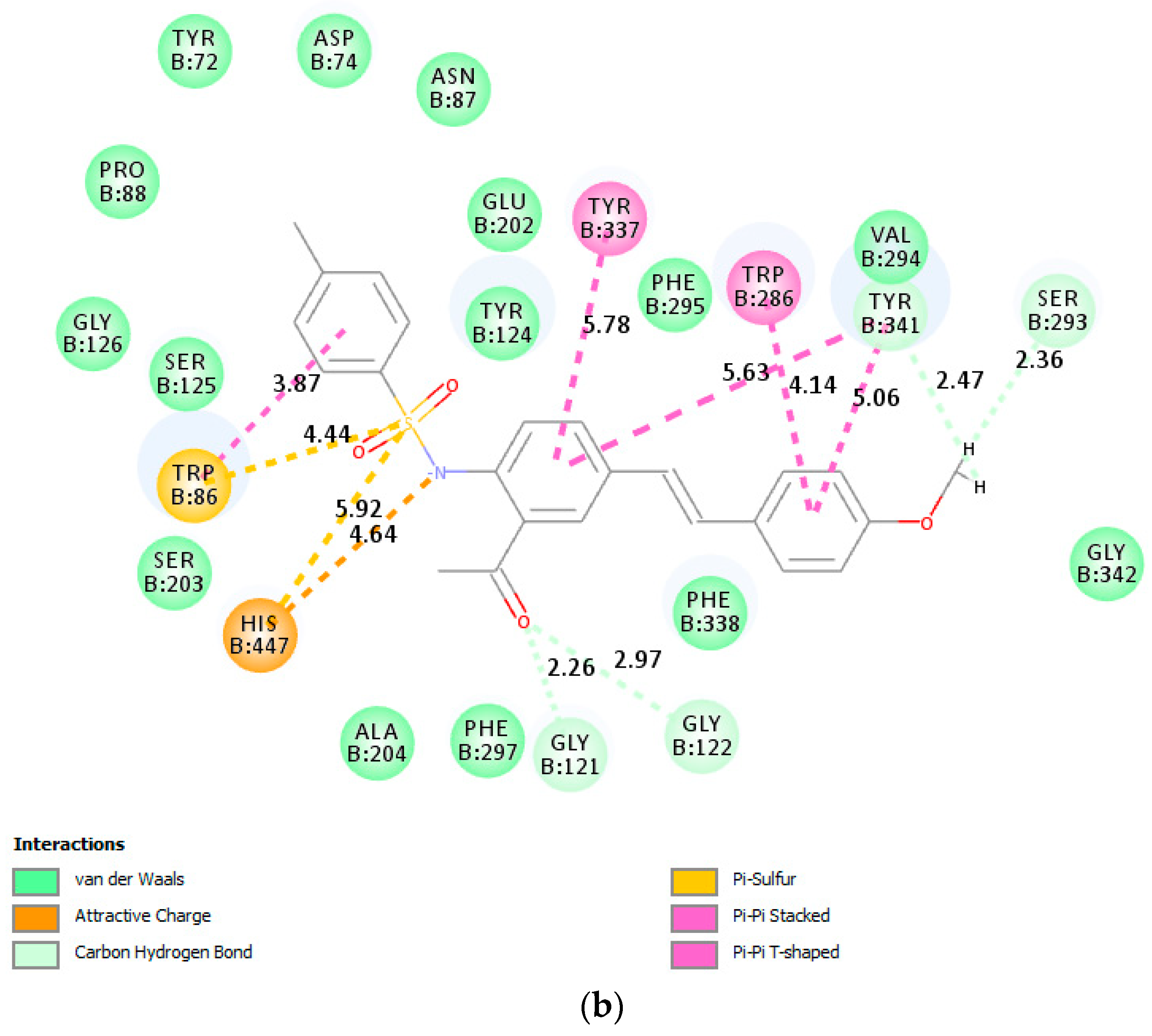
Donepezil and compound
3a were also docked into the active site of BChE crystal (PDB code: 1P0I). The top scoring docked pose of donepezil (
Figure 8a) shows hydrogen bonding interactions between the carbonyl and methoxy oxygen atoms of donepezil with the protein residue Thr120 and Tyr128 of BChE, respectively. Although the conjugated framework of
3a is embedded in the active site of BChE (
Figure 8b), molecular docking predicts no interaction of the 4-methoxystyryl and acetophenone scaffolds with any of the protein residues of the enzyme. However, nitrogen and oxygen atoms of the sulfonamide group are predicted to be involved in carbon hydrogen bonding interactions with His438 of CAS. His438 and Trp82 are involved in π···sulfur interaction with sulfur atom of the sulfonamide moiety. The phenyl ring and methyl group of the 4-tosylamino group are involved in π-π stacked and alkyl interactions with Trp82.
The drug-likeness of the test compounds was predicted at theoretical level through the Lipinski rule of five which states that for a molecule to act as a therapeutic candidate it must possess four characteristics, which are (i) number of hydrogen bond donors should not be more than 5, (ii) number of hydrogen bond acceptors should not be more than 10, (iii) the molecular mass should be less than 500 Da, and (iv) LogP (octanol-water partition coefficient) should not be greater than 5. Small drug-like molecules with moderate polarity (PSA < 79 Å) and lipophilicity (logP from + 0.4 to + 6.0) have a high probability to cross the BBB by passive diffusion to reach the CNS [
49]. For CNS agents or drugs to permeate the BBB, the molecules should be lipophilic and have smaller sizes (less than 500 Dalton) to be able to pass through the BBB [
27]. Compounds
3a and
3b (
Table 3) with LogP greater than 5 violate the Lipinski rule and may not be able to cross the BBB. It has been reported that excessive hydrogen bond donors and acceptors increase the polarity of the molecule, in turn, reduce permeability across the BBB [
27].














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}