1. Introduction
Throughout the last 20 years the SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) surfaces have been broadly explored theoretically and experimentally [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10]. At the same time, to the best of our knowledge, there are no reports of ab initio calculations dealing with the atomic relaxation and electronic structure of the pristine WO
2-terminated WO
3 (001) surface in the cubic perovskite-like structure. Nevertheless, a large amount of experimental studies exist dealing with WO
3 (001) surfaces [
11,
12,
13,
14]. Recent theoretical studies have been devoted, for example, to hydrogen adsorption on the WO
3 (001) surface [
15], understanding the water splitting process on the WO
3 (001) surface [
16], and H
2O adsorption on the WO
3 (001) surface [
17].
BaTiO
3, PbTiO
3 and CaTiO
3 perovskites have attracted huge fundamental interest in these materials mostly for their phase transitions. Historically the ABO
3 perovskites were highly promising low-cost energy materials. They have been used for numerous optoelectronic and photonic device applications [
18]. SrTiO
3 perovskite thin films are important for a large amount of technologically important applications [
19,
20]. For example, they are used for catalysis, optical wave guides, high-capacity memory cells as well as substrates for high-temperature cuprate superconductor growth [
19,
20]. Barium titanate (BaTiO
3) is an excellent photorefractive material [
18]. Ferroelectric PbTiO
3 thin films have been applied to large numbers of electronic devices, such as non-volatile memory FET [
19] and Si monolithic ultrasonic sensors [
18]. CaTiO
3 is used worldwide in technologically important electronic ceramic materials [
18]. Tungsten trioxide (WO
3) and its thin films exhibit a large number of novel properties useful for high-technology applications [
21]. In particular, WO
3 undergoes phase transitions, which are explored for their potential in industrial applications, display systems and microelectronics [
21]. It is worth noting that the predictive power of ab initio calculations makes possible the design of new materials for high-technology applications on paper. Nowadays, consumer electronics mostly use lithium-ion batteries containing LiCoO
2 cathode, which was discovered in 1980 by J. Goodenough, one of the 2019 Nobel Prize winners for Chemistry [
22]. The experimentally detected LiCoO
2 average intercalation voltage is 4.0–4.1 V [
23]. Based on ab initio calculations by Eglitis and Borstel [
24,
25,
26], it was demonstrated that a novel Li
2CoMn
3O
8 battery cathode material can lead to a high-energy lithium-ion battery working at the 5 V regime.
The SrTiO3, BaTiO3, PbTiO3 and CaTiO3 perovskite cubic unit cells contain five atoms. The A type atom (A = Sr, Ba, Pb or Ca) has the coordinates (0, 0, 0), and it is located in the cube corner position. The Ti atom has the coordinates (½, ½, ½), and it is located in the cube body center position. The 3 O atoms have the coordinates (½, ½, 0), (½, 0, ½), (0, ½, ½), and they are located in the cube face centered positions. All SrTiO3, BaTiO3, PbTiO3 and CaTiO3 cubic perovskites have the same space group Pm3m with the space group number equal to 221. WO3 in its cubic perovskite-like structure has exactly the same space group as ATiO3 perovskites Pm3m, and also the same space group number 221. The only striking difference between the SrTiO3, BaTiO3, PbTiO3 and CaTiO3 cubic perovskites as well as WO3 in its cubic perovskite-like structure is that WO3 has an empty A cation position. Thereby, the cubic perovskite-like unit cell of WO3 contain only four atoms.
The objective of the reported here work was to carry out first-principles calculations for WO2-terminated polar WO3 (001) surfaces in the cubic perovskite-like structure. We compared our WO2-terminated WO3 (001) surface-atomic and electronic-structure ab initio calculations with our results for the related structure TiO2-terminated SrTiO3, BaTiO3, PbTiO3 and CaTiO3 cubic perovskite (001) surfaces. We carefully compared our calculation results for all five of our calculated materials and detected systematic common trends. The results for WO2-terminated WO3 and TiO2-terminated SrTiO3, BaTiO3, PbTiO3 and CaTiO3 (001) surfaces were summarized and analysed in a way easily readable for a broad audience of scientists.
2. Computational Methods and Surface Models
In order to carry out ab initio DFT-B3LYP or DFT-B3PW calculations, we employed the CRYSTAL computer program package [
27]. Unlike the plane-wave codes widely employed in many previous studies [
28,
29], the CRYSTAL code [
27] uses localized Gaussian-type basis sets. In our calculations, we adopted the basis sets (BS) developed for SrTiO
3, BaTiO
3 and PbTiO
3 in [
30]. The Hay–Wadt small-core, effective-core pseudopotentials (ECP) were adopted for Ca and Ti atoms [
31,
32,
33]. The small-core ECPs replaced only the inner-core orbitals, while orbitals for subvalence electrons as well as for valence electrons were calculated self-consistently. Oxygen atoms were treated with the all-electron BS. Finally, for the W atom we used BS developed by Cora et al. [
34]. Our calculations were performed by means of the B3LYP [
35] or B3PW [
36,
37,
38] hybrid exchange–correlation functionals. For all WO
3, SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 materials we performed the reciprocal space integration with an 8 × 8 × 8 and 8 × 8 × 1 extension of Pack–Monkhorst mesh for the bulk and (001) surfaces of these materials. The CRYSTAL computer program package [
27] makes possible the calculation of isolated 2D slabs perpendicular to the
Oz direction. In order to compare the performance of different exchange–correlation functionals and choose the best method for our calculations, we calculated the SrTiO
3, SrZrO
3, BaZrO
3, MgF
2 and CaF
2 bulk Γ–Γ band gaps [
30,
39,
40,
41,
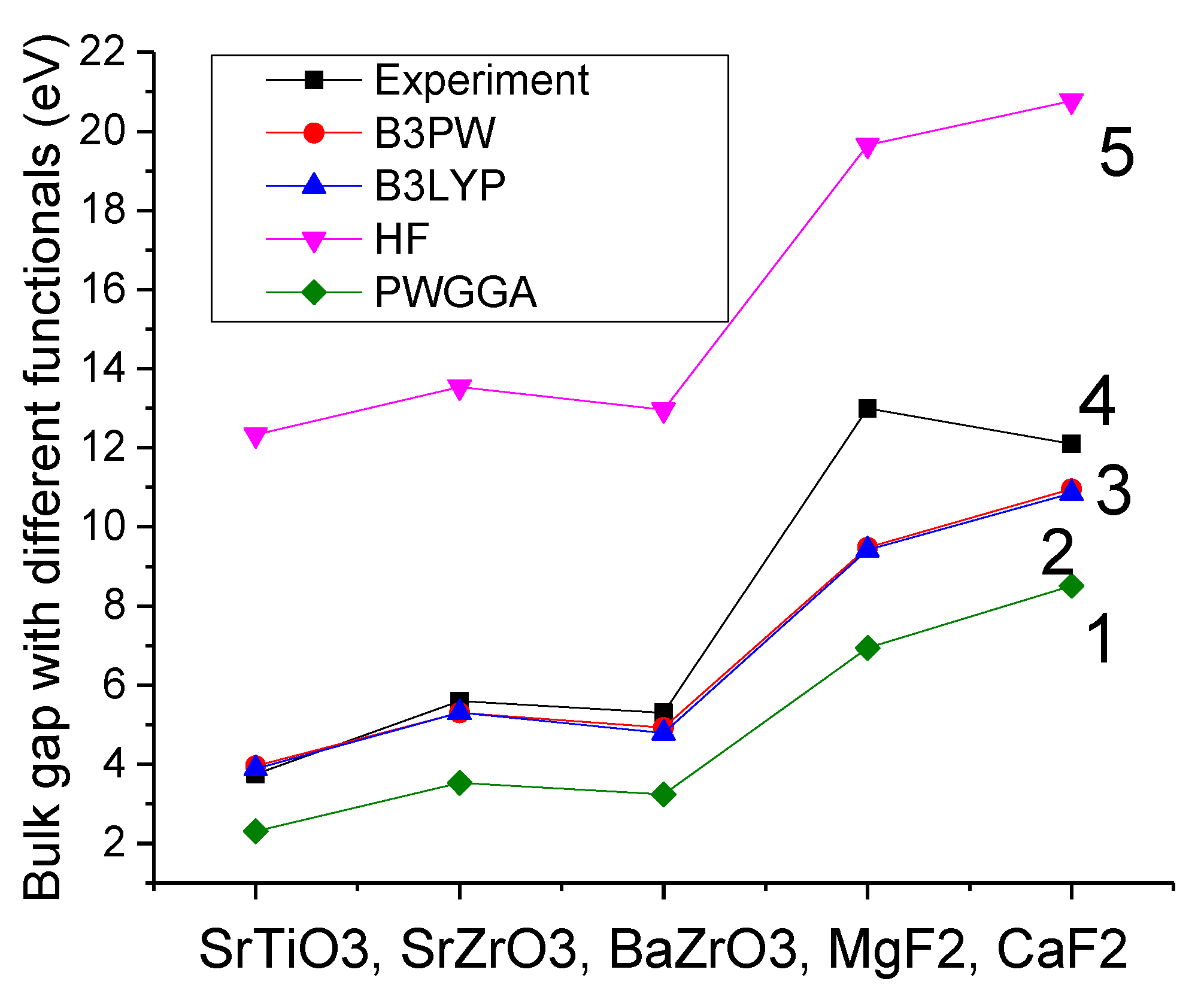
42] (
Table 1 and
Figure 1). The experimentally detected SrTiO
3, SrZrO
3, BaZrO
3, MgF
2 and CaF
2 bulk band gaps at the Γ-point are mentioned in
Table 1 for comparison purposes as well as depicted in
Figure 1 [
43,
44,
45,
46,
47].
As can be seen in
Table 1, the ab initio Hartree-Fock (HF) calculations, for all five our calculated materials, very strongly overestimate the experimental band gap at Γ-point. Namely, the HF method most strongly (3.29 times) overestimate the experimental SrTiO
3 bulk Γ–Γ band gap. Even HF calculated MgF
2 bulk Γ–Γ band gap overestimates the experimental value 1.51 times (
Table 1 and
Figure 1).
From another side, as we can see from
Table 1 and
Figure 1, the generalized gradient approximations (GGA) to the density functional theory (DFT) systematically and considerably underestimate the experimental Γ–Γ bulk band gap in our calculated ABO
3 perovskites as well as MgF
2 and CaF
2. For example, the PWGGA (6.94 eV) and PBE (6.91 eV) calculated MgF
2 bulk band gap at Γ-point is 1.87 and 1.88 times, respectively, smaller than the experimental MgF
2 bulk Γ–Γ band gap value of 13.0 eV [
37].
To obtain the best possible results, we performed our WO
3, SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 bulk and (001) surface calculations by means of the B3PW [
36,
37,
38] or B3LYP [
35] hybrid exchange–correlation functionals. The hybrid functional incorporates a portion of exact exchange energy density from HF theory (20%) while the rest of the exchange–correlation part is a mixture of different approaches (both exchange and correlation). It is obvious, that the B3PW and B3LYP hybrid exchange–correlation functionals, since they are a superposition of HF and DFT methods as implemented in the CRYSTAL computer code [
27], allowed us to achieve as good an agreement as possible between the first principles calculated and the experimentally detected Γ–Γ band gaps for WO
3, SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 bulk and their (001) surfaces.
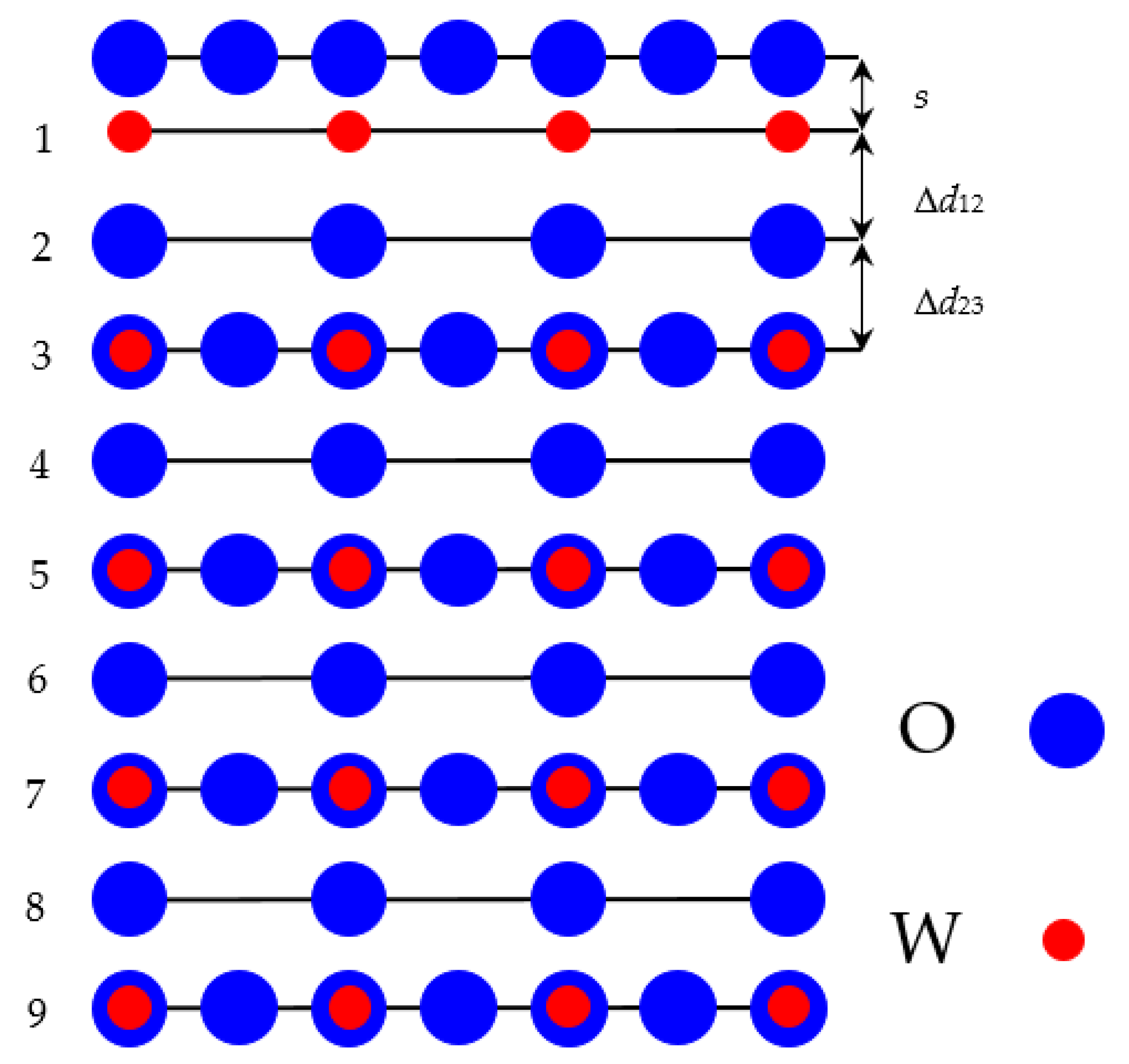
In our ab initio calculations we used WO
2-terminated WO
3 as well as TiO
2-terminated SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) slabs containing 9 alternating layers. First our calculated WO
3 (001) slab was terminated by WO
2 planes from both sides (WO
2–O–WO
2–O–WO
2–O–WO
2–O–WO
2) from a 19-atom supercell (
Figure 2). Another of our calculated SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) slabs was terminated by TiO
2 planes from both sides (TiO
2–AO–TiO
2–AO–TiO
2–AO–TiO
2–AOvTiO
2) and consisted of a 23-atom supercell (
Figure 3). Both our calculated slabs were non-stoichiometric and had unit-cell equations W
5O
14 as well as A
4Ti
5O
14, respectively. To analyse the chemical bonds, effective atomic charges and covalency effects for WO
3 and ATiO
3 perovskite bulk and (001) surfaces, we used the well-known Mulliken population analysis [
48,
49,
50,
51,
52].
3. Ab initio Calculation Results for WO3, SrTiO3, BaTiO3, PbTiO3 and CaTiO3 Bulk
As a starting point, by means of the hybrid B3LYP exchange–correlation functional, we calculated the cubic WO
3 bulk lattice constant (3.775 Å). Our calculated cubic WO
3 constant (3.775 Å) was only slightly larger than the experimental value of
a0 = 3.71–3.75 Å [
51] (
Table 2); nevertheless, it was in almost perfect agreement with the earlier calculation result for the WO
3 cubic structure bulk lattice constant calculated by the full-potential linear muffin-tin (FP-LMTO) code equal to 3.78 Å [
52]. Our B3PW-calculated SrTiO
3 bulk lattice constant (3.904 Å) was only slightly overestimated with respect to the experimental SrTiO
3 bulk lattice constant (3.89 Å) extrapolated to 0 K [
53] (
Table 2). Our ab initio calculation of the BaTiO
3 bulk lattice constant (4.008 Å) was in an outstanding agreement with the experimental value of 4.00 Å [
53,
54,
55]. Our B3PW-calculated PbTiO
3 bulk lattice constant (3.936 Å) [
54,
55,
56] was only 0.86% under the experimental value of 3.97 Å [
57]. Finally, our calculated CaTiO
3 bulk lattice constant (3.851 Å) was 1.17% smaller than the experimentally detected (3.8967 Å) [
58,
59,
60] (
Table 2).
Our ab initio B3LYP-calculated effective atomic charges for the WO
3 bulk were (+3.095
e) for the W atom, and (−1.032
e) for each of the three O atoms (
Table 3). Our B3LYP-calculated effective W atomic charge (+3.095
e) was almost two times smaller than the generally accepted classical ionic charge for the W(+6
e) atom. In addition, our calculated effective atomic charge for the O (−1.032
e) atom was almost two times smaller than the generally accepted O atom classical ionic charge (−2
e). In addition, for the SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 perovskites, our calculated A atomic charges (+1.871
e, +1.797
e, +1.354
e and +1.782
e, respectively) were considerably smaller than those of the classical Sr, Ba, Pb, Ca atom ionic charges (+2
e) (
Table 3) [
61,
62,
63,
64,
65,
66]. Our B3PW-calculated O atom Mulliken charges in SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 perovskites (−1.407
e, −1.388
e, −1.232
e and −1.371
e, respectively) are also at least 29.65% smaller than the classical ionic O atomic charge (−2
e) [
67,
68,
69]. Finally, our ab initio-calculated SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 Ti atomic charges (+2.351
e, +2.367
e, +2.341
e and 2.330
e) are more than one-and-a-half times smaller than the formal Ti atom ionic charge (+4
e). Our calculated chemical bond population between W and O atoms in WO
3 bulk (0.142
e) is approximately one-and-a-half times larger than the Ti–O atom chemical bond population in SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 perovskites (+0.088
e, +0.098
e, +0.098
e and +0.084
e, respectively).
Our B3LYP-calculated WO
3 bulk Γ–Γ band gap (4.95 eV) overestimated by 1.21 eV the experimental direct WO
3 bulk band gap value at Γ-point of 3.74 eV [
70] (
Table 4). Moreover, our B3PW-calculated bulk Γ–Γ band gaps for SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 perovskites (3.96 eV, 3.55 eV, 4.32 eV and 4.18 eV, respectively) were always slightly overestimated with respect to the experimentally measured direct band gap values at Γ-point for SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 perovskites (3.75 eV [
43], 3.2 eV [
71], 3.4 eV [
72] and 3.5 eV [
73], respectively) (
Table 4).
4. Ab Initio Calculation Results for the WO2-Terminated WO3 as Well as TiO2-Terminated SrTiO3, BaTiO3, PbTiO3 and CaTiO3 (001) Surfaces
Our B3LYP- or B3PW-calculated atomic displacements for the WO
2-terminated WO
3 and the TiO
2-terminated SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) surface upper-three or two layers are presented in
Table 5. According to our B3LYP or B3PW calculations, all atoms of the first (upper) surface layer relaxed inwards, while all second-layer atoms relaxed outwards (
Table 5). The only two exceptions to this systematic trend were the outward relaxation of the first layer O atom of the WO
2-terminated WO
3 (001) surface (+0.42% of a
0) and the outward relaxation of the TiO
2-terminated PbTiO
3 (001) surface first-layer O atom by (0.31% of a
0) (
Table 5). The first layer metal atom relaxation magnitudes range from −1.71% of a
0 for the TiO
2-terminated CaTiO
3 (001) surface to −3.08% of a
0 for the TiO
2-terminated BaTiO
3 (001) surface (
Table 5). The first- and second-layer metal atom displacement magnitudes for WO
2-terminated WO
3 and TiO
2-terminated SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) surfaces were always considerably larger than the respective first- and second-layer O atom displacement magnitudes (
Table 5).
Our B3LYP-calculated surface rumpling amplitude
s (the relative displacement of an oxygen atom relative to the metal atom in the upper surface layer) for WO
2-terminated WO
3 (001) surface (+2.49) is in qualitative agreement with our B3PW-calculated surface rumpling amplitudes
s for TiO
2-terminated BaTiO
3, PbTiO
3, CaTiO
3 and SrTiO
3 (001) surfaces (+2.73, +3.12, +1.61 and +2.12, respectively) (
Table 6). Our B3PW-calculated surface rumpling amplitude
s for TiO
2-terminated SrTiO
3 (001) surface (+2.12) is in fair agreement with available RHEED (+2.6 [
74]) and LEED (+2.1 ± 2 [
75]) experimental data (
Table 6). Unfortunately, our B3PW-calculated interlayer distance Δ
d12 for the TiO
2-terminated SrTiO
3 (001) surface (−5.80) had the opposite sign to the experimentally measured RHEED (+1.8 [
74]) and LEED (+1 ± 1 [
67]) interlayer distances (
Table 6). Finally, our B3PW-calculated interlayer distance Δ
d23 for the TiO
2-terminated SrTiO
3 (001) surface (+3.55) is in qualitative agreement with the RHEED experiment result (+1.3), but had the opposite sign to that of the LEED experimental result (−1 ± 1). Nevertheless, it is worth noting that the RHEED (+1.3) and the LEED experiments (−1 ± 1) had opposite signs for the interlayer distance Δ
d23 (
Table 6).
We started the discussion of the electronic structure of WO
2-terminated WO
3 and TiO
2-terminated SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) surfaces with an analysis of charge redistribution in the top-three surface planes (
Table 7). The ab initio-calculated atomic displacements, bond populations between the nearest metal and oxygen atoms and the effective atomic charges are collected in
Table 7. For example, the effective static atomic charges on WO
2-terminated WO
3 as well as TiO
2-terminated SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) surface upper-layer W and Ti atoms are always reduced in comparison to the bulk WO
3, SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 crystal charges (−0.312
e, −0.06
e, −0.06
e, −0.062
e and −0.052
e, respectively). We recently observed a similar effect: the reduction of surface upper-layer metal atomic charges near the ReO
2-terminated ReO
3 and the ZrO
2-terminated SrZrO
3, BaZrO
3, PbZrO
3 and CaZrO
3 (001) surfaces [
76]. According to our ab initio calculations, the largest upper-layer metal atom displacement was observed for the TiO
2-terminated BaTiO
3 (001) surface Ba atom (−0.123 Å). Nevertheless, the TiO
2-terminated PbTiO
3 (001) surface second-layer Pb atom outward displacement (+0.209 Å) was even larger.
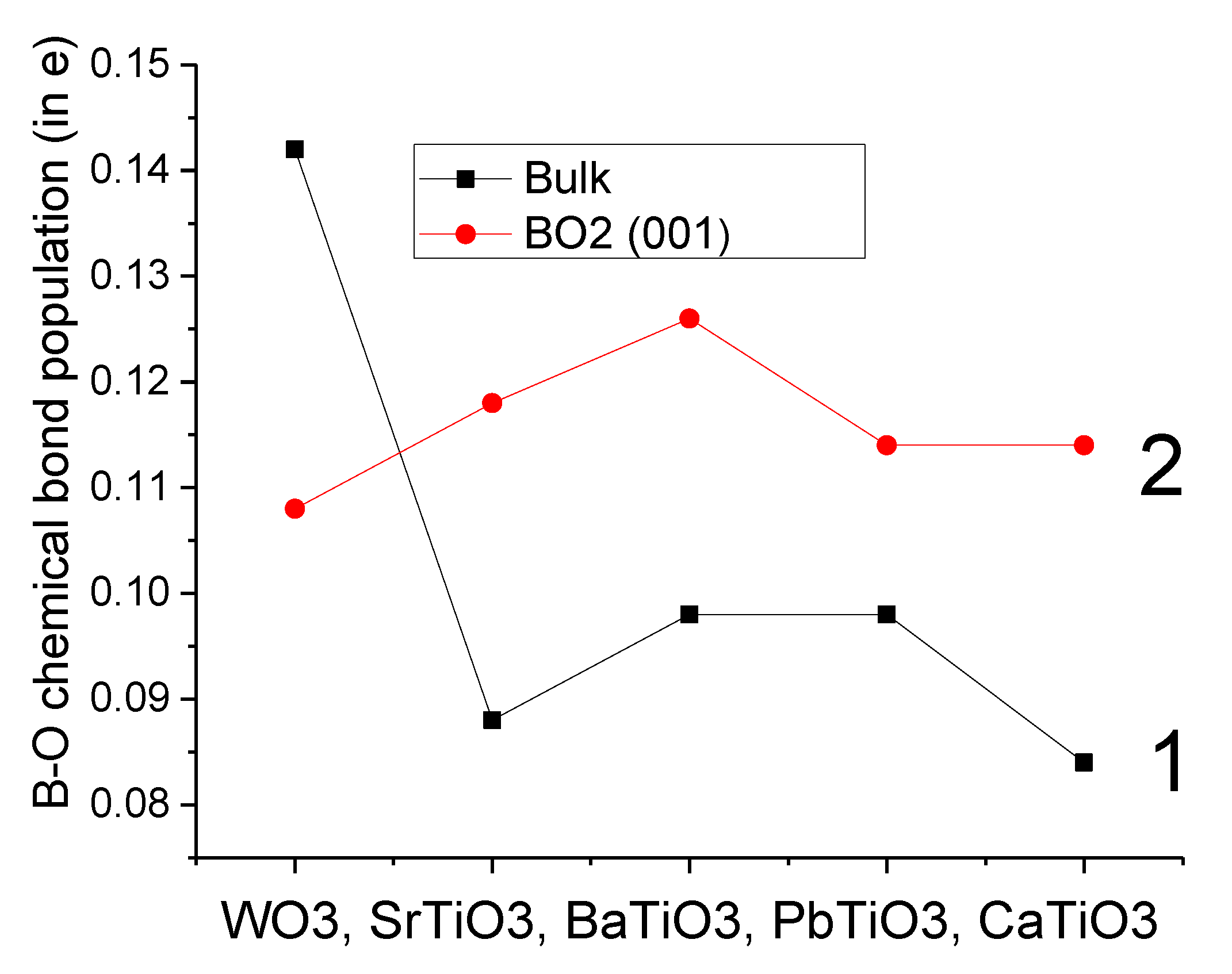
Our B3PW-calculated SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 bulk Ti-O chemical bond covalency (+0.088
e, +0.098
e, +0.098
e and +0.084
e, respectively) were always smaller than near the TiO
2-terminated SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) surfaces (0.118
e, 0.126
e, 0.114
e, 0.114
e, respectively) (
Table 8 and
Figure 4). Just opposite situation was obtained for the WO
3 crystal: the B3LYP-calculated W-O chemical bond population in the WO
3 bulk (0.142
e) was larger than near the WO
2-terminated WO
3 (001) surface (0.108
e) (
Table 8 and
Figure 4). Nevertheless, it is worth noting that the W–O chemical bond population between the W atom on the top layer of WO
2-terminated WO
3 (001) surface and the O atom on the second layer (0.278
e) is the largest one (
Table 8), which was in agreement with our previous B3LYP calculations dealing with ReO
2-terminated ReO
3 (001) surfaces [
76].
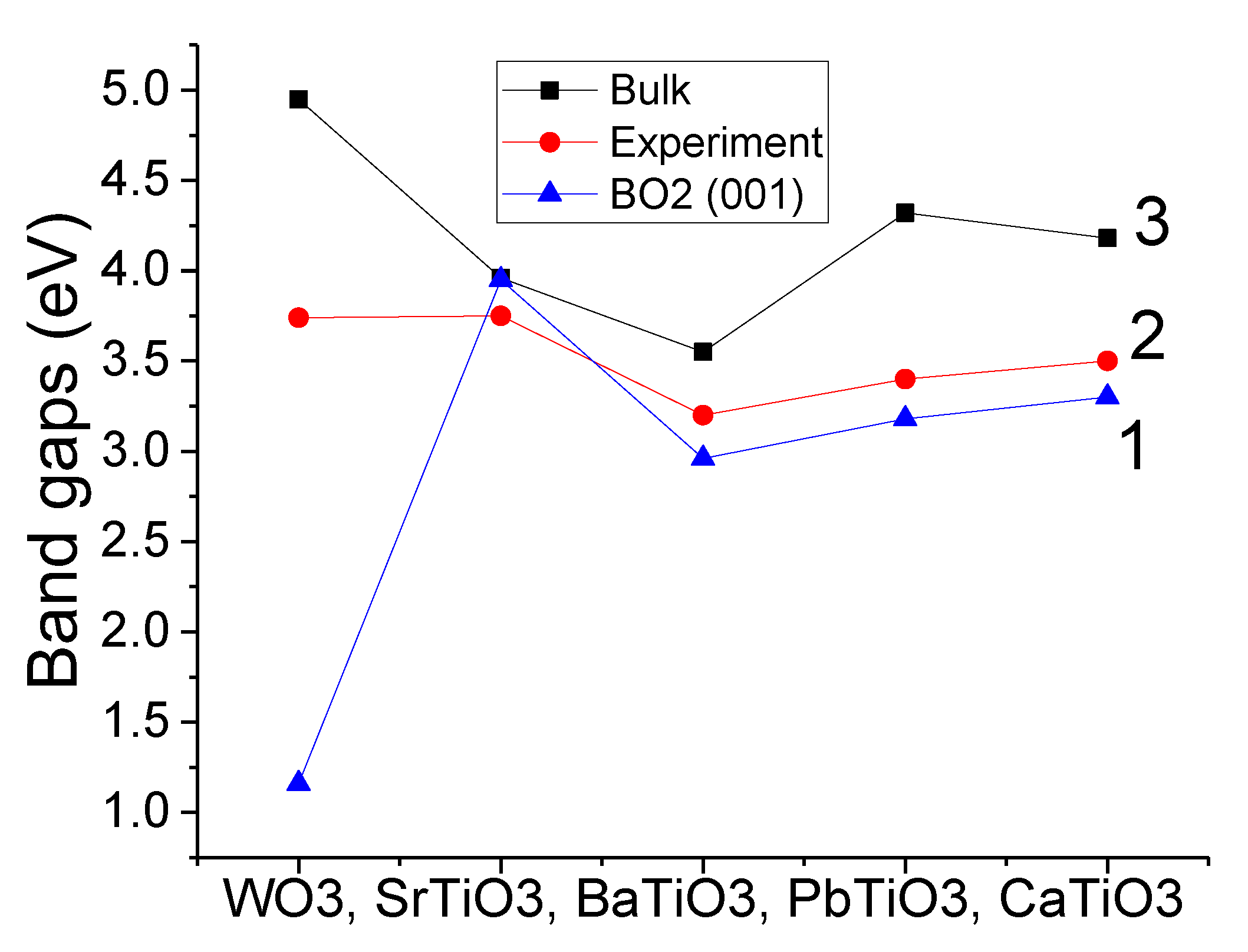
As can be seen in
Table 9 and
Figure 5, our B3LYP- or B3PW-calculated WO
3, SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 bulk band gaps at the Γ–Γ point were always reduced near the WO
2- or TiO
2-terminated WO
3, SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) surfaces. The B3PW-calculated SrTiO
3 bulk band gap at Γ–Γ point near the TiO
2-terminated SrTiO
3 (001) surface at Γ–Γ point was reduced only by 0.01 eV. At the same time, our B3LYP-calculated WO
3 bulk band gap (4.95 eV) at the Γ–Γ point near the WO
2-terminated WO
3 (001) surface was reduced by 3.79 eV to 1.16 eV (
Table 9 and
Figure 5).
5. Conclusions
For the first principles-calculated WO
2- or TiO
2-terminated WO
3, SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) surfaces, as a rule, all first-layer surface atoms relax inwards, whereas all second-layer surface atoms relax upwards. The only two exceptions from this systematic trend are the upward relaxation of WO
2- or TiO
2-terminated WO
3 and PbTiO
3 (001) surface first-layer O atoms. As a result of our ab initio-calculated atomic relaxation, TiO
2-terminated SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) surfaces exhibited a reduction of the interlayer distance Δ
d12 (−5.80, −5.59, −8.13, −4.46% of
a0, respectively) as well as an expansion of Δ
d23 (+3.55, +2.51, +5.32, +2.75% of
a0, respectively). It is worth noting that after geometry optimization, it is very useful to perform ab initio molecular dynamics computations to ensure the stability of the structures over time [
77].
The changes in the interlayer distances between the first and second layer (Δd12) were always larger than between the second and third layer (Δd23) for all our calculated perovskites SrTiO3, BaTiO3, PbTiO3 and CaTiO3.
The Ti–O chemical bond population in SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 bulk was always smaller than near their TiO
2-terminated (001) surface (see
Figure 4). In contrast, the W–O chemical bond population in the WO
3 bulk (0.142
e) was larger than near the WO
2-terminated WO3 (001) surface (0.108
e). Nevertheless, the largest W–O chemical bond population, according to our ab initio calculations, is between the W atom located on the WO
2-terminated WO
3 (001) surface and the second-layer O atom (0.278
e). It was worth noting, that also for the related material ReO
3, according to our calculations [
76], the situation was similar. Namely, the Re–O chemical bond population in the ReO
3 bulk (0.212
e) was larger than near the ReO
2-terminated ReO
3 (001) surface (0.170
e). Nevertheless, the Re–O chemical bond population between the Re atom located on the ReO
2-terminated ReO
3 (001) surface upper-layer and O atom located on the ReO
2-terminated ReO
3 (001) surface second layer was the largest (0.262
e).
According to our B3LYP or B3PW calculations, the WO
3, SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 bulk Γ–Γ band gap values (4.95, 3.96, 3.55, 4.32, 4.18 eV, respectively) were always reduced with respect to the bulk near the WO
2- or TiO
2-terminated WO
3, SrTiO
3, BaTiO
3, PbTiO
3 and CaTiO
3 (001) surfaces (1.16, 3.95, 2.96, 3.18, 3.30 eV, respectively) (see
Figure 5).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}