1. Introduction
In December of 2019, the first cases of a new respiratory disease were reported in Wuhan, China. The disease rapidly spread globally, prompting the World Health Organization (WHO) to announce a worldwide pandemic. The term Coronavirus Disease 2019 (COVID-19) was assigned to the pandemic, and its causative agent was identified as a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [
1,
2,
3]. The COVID-19 virus is a new member of the Betacoronavirus genus, which includes severe acute respiratory syndrome-related coronavirus (SARS-CoV), the Middle East respiratory syndrome coronavirus (MERS-CoV), and various bat coronaviruses [
4]. SARS-CoV-2 has been linked to an elevated risk of human-to-human transmission. This is demonstrated by the finding that the SARS-CoV-2 spike protein has a greater affinity for human host cells [
5,
6,
7].
SARS-CoV-2 is classified as a positive-sense RNA virus. The virus encodes 16 non-structural proteins (nsps) to assemble its replication and transcription complex (RTC), which facilitates virus replication and transcription. Specifically, nsp12 functions as an RNA-dependent RNA polymerase (RdRp) with nsp7 and nsp8 as cofactors. COVID-19 replication and transcription are mediated by the RdRp, which catalyzes viral RNA production; thus, the RdRp plays a crucial role in the survival and replication of the virus [
8,
9,
10]. The active RdRp site is highly conserved, with two successive and surface-accessible aspartates in a beta-turn structure [
11]. Several new antiviral drugs targeting the RdRp domain, such as the nucleotide analog antiviral inhibitor remdesivir, have shown promising results against SARS-CoV-2. Remdesivir is a prodrug that is converted to the active triphosphate form in the body [
12,
13].
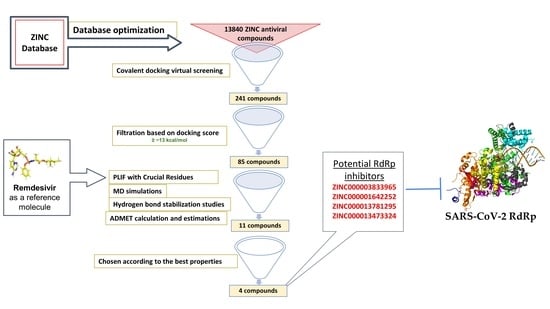
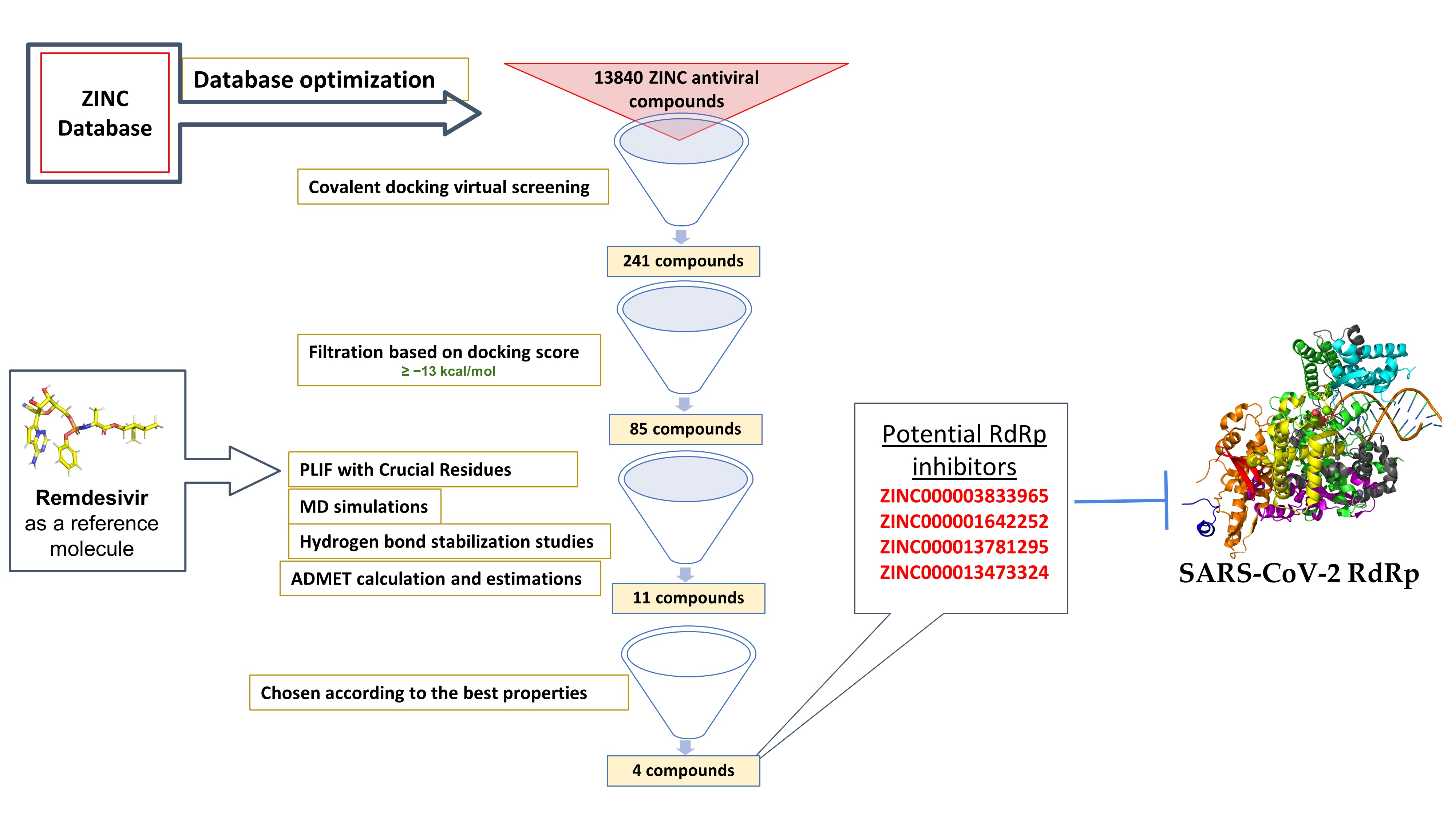
With the current pace of infection and the high mortality rate of SARS-CoV-2 worldwide, researchers are investigating possible treatment mechanisms to address the crisis and halt the virus’s spread. One pioneer method in drug discovery is in silico drug discovery and design. Computer-aided drug design allows researchers to explore possible mechanisms for use in disease treatment. In this report, we utilize computer-aided techniques, such as virtual screening, molecular docking, and molecular dynamics (MD) simulations, alongside additional tools to predict pharmacokinetic and toxicity parameters and identify potential new covalent inhibitors of SARS-CoV-2 RdRp from the antiviral ZINC database. The newly discovered inhibitors could pave the way for designing new molecules targeting the SARS-CoV-2 RdRp
2. Material and Methods
2.1. Database Preparation
A library consisting of approximately 13,840 compounds was retrieved in SDF format from the ZINC database website [
14]. For structure-based virtual screening, the prepared library included FDA-approved drugs that act against viral proteases as well as antiviral compounds from natural and synthetic sources. Then, the SDF files were converted to MDB format using MOE 2015.0101 (Molecular Operating Environment, Chemical Computing Group Inc., Montreal, Quebec, Canada) to form the MOE database. Remdesivir (2-ethylbutyl (2
S)-2-[[[(2
R,3
S,4
R,5
R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxyoxolan-2-yl]methoxyphenoxyphosphoryl]amino]propanoate) was downloaded from PubChem (ID: 121304016) for use as a reference molecule. Ligands were prepared by adding hydrogens to all compounds, after which energy minimization was performed using the MMFF94x force field. Furthermore, ligand geometries were optimized using a root mean square (RMS) gradient of 0.05 kcal/mol/Å. The optimized ligands were exported in MDB format for further processing.
2.2. Structure-Based Virtual Screening
The SARS-CoV-2 RdRp model (PDB ID: 7BV2, chains A, B, and C) [
15] was used for molecular docking. The polyprotein replicate 7BV2 has a resolution of 2.50 Å and cleaves the C-terminus at 11 sites. The known ligand of 7BV2 boceprevir was used as a control. To improve the compound selectivity in this study, all hits previously obtained at the SARS-CoV-2 binding site of RdRp were docked using the covalent docking protocol in MOE. Therefore, the covalent docking protocol was validated by redocking of a native ligand (remdesivir) before beginning the study, for which the resulting root mean square deviation (RMSD) value was less than 2 Å.
Covalent docking studies with the designed database were performed using MOE version 2015.0101 (Chemical Computing Group Inc., Montreal, QC, Canada) by setting the primer strand at the first replicated base pair as the site of the covalent bond in the RdRp enzyme and applying a Michael addition reaction. The following parameters were set as the docking calculi: the residues were kept as flexible residues, the number of refinements of retained poses was set to 100, and the number of generated conformations was set to 10. Final pose scoring was performed based on the London dG free binding energy. In addition, the generalized-born volume integral/weighted surface area (GBVI/WSA) free binding energy rescoring function was utilized to rescore the final poses. For further analysis of the protein–ligand interactions, the docked poses with the lowest London dG values were chosen.
During the docking process, protonation of the protein was performed by the Protonate 3D method [
16], and energy minimization was performed using the MOE-implemented Amber12 force field. To specifically identify the protein’s active site, the grid was set to include the ligands in the PDB file. During docking, the triangular matching algorithm placement method was used in addition to the London dG scoring function. Furthermore, for the rescoring function, the GBVI/WSA dG was utilized. All compounds were docked in the given binding site. Next, the protein–ligand interaction fingerprinting (PLIF) module in MOE was applied to fingerprint the binding interactions of crucial residues between the compounds and the protein. Additional associations and poses of binding were visually studied using MOE [
17]. Based on the binding profiles, 11 hit compounds from the ZINC database were subjected to MD simulation studies.
2.3. Molecular Dynamics Simulations
The NAMD 2.13 suite (
http://www.ks.uiuc.edu/Research/namd; accessed on 21 May 2020) and MOE were employed for the molecular dynamics simulations [
18]. Several reports involving MD simulations, including protein–ligand interaction studies, have documented using this approach [
19]. Using the solvation algorithm in MOE, the structures were immersed in a TIP3P water box extending 10 Å from all protein atoms. Additionally, the counter ions Na
+ and Cl
− were added to ensure charge neutrality, but the ion concentration was maintained at or below 0.15 M. System equilibration was carried out using a conjugate gradient energy minimization composed of 10,000 steps. A system warm-up was conducted from 50 K to 310 K with 0.1 ns at each temperature increment, followed by equilibration for 0.1 ns at a fixed temperature of 310 K. Next, an unrestricted run lasting 10 ns was performed, succeeded by cooling from 300 K to zero over 0.1 ns. The time step was set as 2 fs, and the particle mesh Ewald method was utilized to evaluate the long-range electrostatic interactions. The Visual Molecular Dynamics (VMD) 1.9.3 (the Theoretical and Computational Biophysics group, NIH Center for Macromolecular Modeling and Bioinformatics, at the Beckman Institute, University of Illinois at Urbana-Champaign, Champaign, IL, USA) analysis tool was used to assess the trajectories. To gain insight into the stability of each complex between the protein and the ligand, the following parameters were evaluated during the MD simulation: RMSD, root mean square fluctuation (RMSF), and radius of gyration (R
oG).
2.4. Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Studies
In drug development and design, analysis of several pharmacokinetic parameters is essential for the development of drug-like molecules. Prediction of the ADMET properties was performed utilizing the ADMET descriptors in Discovery Studio 4.5 (Accelrys, San Diego, CA, USA) [
20]. The module used mathematical models to quantitatively predict the corresponding properties. To predict human intestinal absorption (HIA) after oral administration, the ADMET absorption model was used. Based on 2D polar surface area (PSA_2D) and AlogP (ADMET_AlogP98) measurements, the model was constructed using 199 compounds in the training set. The HIA model is defined by 95% and 99% confidence ellipses in the ADMET_PSA_2D and ADMET_AlogP98 plane, respectively [
21]. The upper limits of the PSA_2D value for the 95% confidence ellipsoid and the PSA_2D value for the 99% confidence ellipsoid were set as 131.62 and 148.12, respectively. To predict the compound water solubility at ambient temperature (25 °C), the ADMET aqueous solubility model was used. Using a set of 784 compounds with known aqueous solubilities in the training set, the model employs the genetic partial least squares (PLS) process for calculations [
22]. In addition, the blood–brain barrier (BBB) permeability of a molecule was predicted by the ADMET BBB model. A quantitative linear regression model was used to derive the predicted blood–brain penetration of the discovered compounds by referencing over 800 compounds known to be BBB-permeable after oral administration. In the ADMET PSA 2D-ADMET AlogP98 plane, 95% and 99% confidence ellipses were also predicted [
23]. The ADMET plasma protein binding model was utilized to predict each compound’s plasma protein binding ability. The model is based on AlogP98 and 1D similarities to two sets of marker molecules. The two marker sets were flagged based on a set binding level. The binding levels for marker flagging were set at 90% or greater binding and 95% or greater binding. The predicted binding levels were modified according to the calculated logP [
24]. Cytochrome P450 2D6 enzyme inhibition was predicted using the 2D chemical structure as input and the probability estimate for the prediction. A training set of 100 compounds with known CYP2D6 inhibition activities was used for the predictions [
24]. Finally, potential human hepatotoxicity prediction was performed based on 382 known hepatotoxic training compounds [
25].
3. Results and Discussion
3.1. Inhibitor Binding Cleft of RdRp
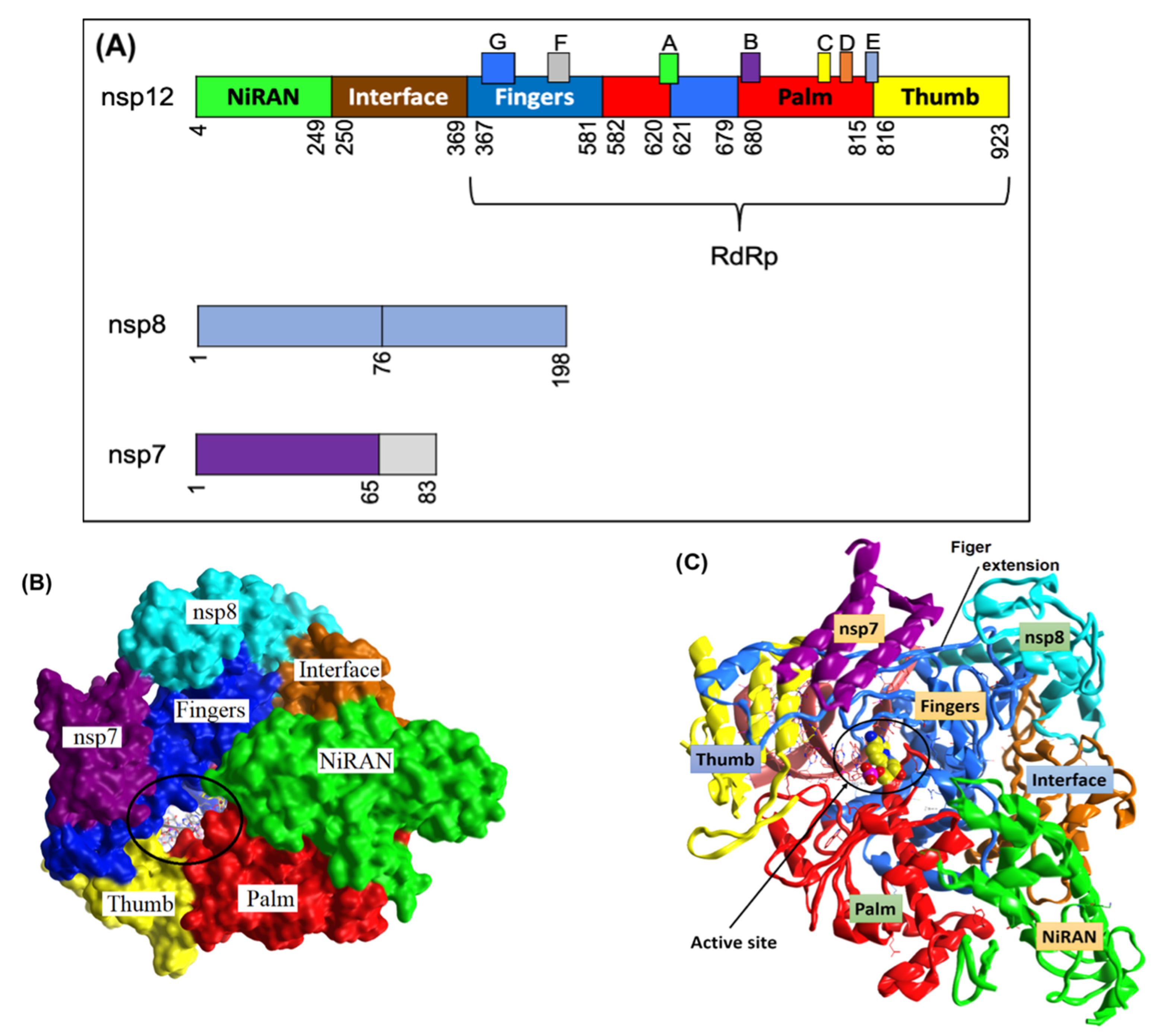
The RTC of SARS-CoV-2 is composed of 16 nsps. Open reading frame-1a (ORF1a) and ORF-1ab viral polyproteins encode these nsps, which aid the replication and transcription of the virus [
26]. Among these nsps, the catalytic subunit nsp12 functions as RNA-dependent RNA polymerase by utilizing nsp7 and nsp8 as cofactors. Therefore, nsp12 plays a crucial role in the replication and transcription cycle of the COVID-19 virus (
Figure 1A) [
26].
To gain insight into the structure and function of SARS-CoV-2 nsp12, the structure of the nsp12–nsp7–nsp8 complex was solved at a 3.7 Å resolution. As a result, the main-chain trace and the bulkiest side chains of each subunit were resolved [
27]. However, the density map did not address the
N-terminal of nsp12 (about 110 amino acids), the
N-terminal of nsp8 (about 80 amino acids), nor a small part of the nsp7 C-terminus (
Figure 1B). Importantly, over 80% structural interpretation was achieved for the 160 kDa protein complex.
The polymerase complex comprises the nsp12 core catalytic subunit linked to the nsp7–nsp8 heterodimer and an additional nsp8 subunit bound at a different binding site (
Figure 1B,C) [
27]. The nidovirus RdRp-associated nucleotidyltransferase (NiRAN) domain is located at the
N-terminal of the nsp12 polymerase subunit. The NiRAN domain is shared by all
Nidovirales members [
28]. The back side of the right hand-configured C-terminal RdRp links to the NiRAN domain. The interface domain linking the NiRAN domain to the finger subdomain of RdRp is between the NiRAN domain and the C-terminal RdRp (
Figure 1). In addition, it has been shown that NiRAN and the interface domain are crucial structural features of the coronavirus RdRp, especially compared to other positive-sense RNA viruses [
29].
The C-terminal catalytic domain is composed of the finger, palm, and thumb subdomains, which are considered conserved structural features of all viral RdRps (
Figure 1). Another unique feature of the SARS-CoV-2 RdRp is the closed-ring structure formed by the long finger extension intersecting with the thumb subdomain (
Figure 1). In contrast, a smaller loop is formed in segmented negative-sense RNA virus (sNSV) polymerases, resulting in an open-conformation structure [
30]. For positive-sense RNA viruses, closed contact between the fingers and the thumb subdomain is a typical structural feature [
31,
32].
3.2. Structure-Based Virtual Screening
In this study, we performed structure-based virtual screening with an antiviral compound database against the viral polymerase using the crystal structure of the RdRp of SARS-CoV-2. This method computationally fit the database molecules into the 3D structure of the active site. Then, the resulting interaction profiles were analyzed to rank the molecules based on the highest binding interactions. The crystal structure of the SARSCoV-2 RdRp was recently solved by Yin, Wanchao et al. (PDB ID code 7BV2) [
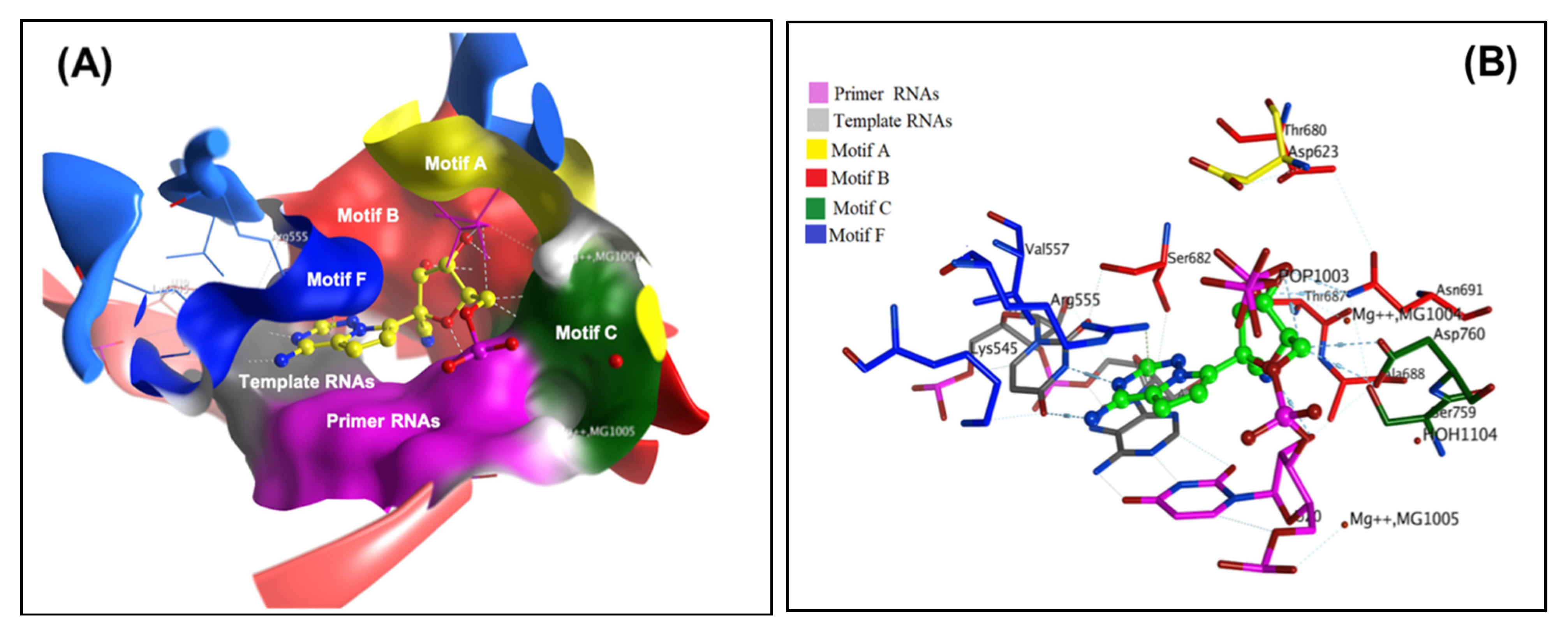
33]. As this crystal structure represents an element necessary for viral replication and survival, it was utilized as the initial point for our computational studies. The arrangement of the SARS-CoV-2 nsp12 catalytic domain follows the typical right-hand configuration exhibited by all viral RdRps. As seen in
Figure 1A and
Table 1, seven critical motifs (A–G) have been reported within the RdRp catalytic domain. Specifically, motifs A–F are strongly conserved across all viral RdRps, while motif G is a unique structural feature of primer-dependent RdRps found in some positive-sense RNA viruses, where it is involved in RNA synthesis initiation by interacting with the primer strand (
Figure 2). Motif C resides in a β-turn loop linking two adjacent strands and contains the essential catalytic residues Ser759–Asp760 and Asp761. By forming a fingertip that extends into the catalytic chamber, motif F interacts with the finger extension loops and the thumb subdomain (
Figure 1A).
The ZINC antiviral compound library was utilized for structure-based virtual screening against the crystal structure of the coronavirus RdRp of SARS-CoV-2. The process computationally fit suitable molecules into the 3D structure of the active site of the target protein. Next, these compounds were ranked according to their interaction profiles. Each interaction profile was studied via covalent docking carried out using MOE 2015 and the SARS-CoV-2 RdRp (PDB ID: 7BV2) [
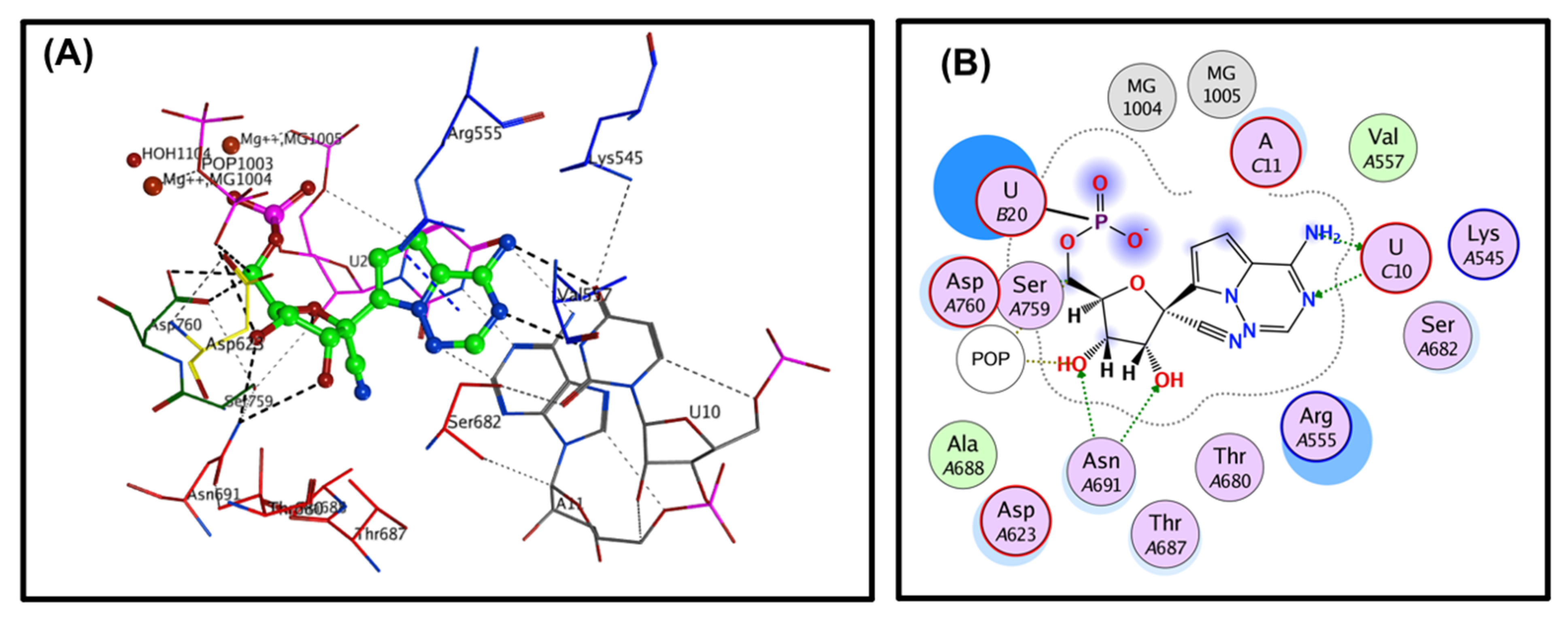
33]. The covalent binding site of RdRp, which was described previously, contains crucial conserved catalytic residues Ser759, Asp760, and Asp761, along with other crucial residues, i.e., Ser549, Lys551, Arg555, Thr190, Cys813, Ser814, and Gln815. The docking process used to filter the prepared database was based on the potential chemical probes that exhibited a higher binding affinity for the active site residues described above. To ensure that all important active site residues were considered, the control molecule Remdesivir was used as a template (
Figure 3). Approximately 13,840 compounds were covalently docked, of which 241 irreversibly interacted and exhibited significant affinities, with varied docking scores (all docking scores were less than −13.0).
The PLIF module in MOE was used to analyze the interactions between the residues of the active site and the hit molecules. The results of the candidate compounds indicated hydrogen bond interactions with crucial residues. Therefore, from 241 initial compounds, 85 hits were chosen according to significant interactions with the catalytic dyad and other hotspot residues of the active site. The 2D molecular docking interactions of the 11 most potent compounds among the 85 identified were specifically analyzed, as seen in
Table 2. The top hits were also selected for MD simulations. The results of docking interactions indicated that among the drugs, compound ZINC000014944915 exhibited the highest binding affinity (−20.37 kcal/mol), as shown in
Table 2 and
Table S1 (Supplementary Materials).
The 11 selected compounds displayed significant interactions with the active site (Ser759, Asp760, and Asp761). The ZINC antiviral database contains compounds with known antiviral activity as well as other biological activities. For example, the hit compounds ZINC000027556215, ZINC000013556344, ZINC000003589958, ZINC000001642252, ZINC000028525778, ZINC000027557701, and ZINC000013781295 have been reported to have antiviral activity against human immunodeficiency virus-type1 (HIV-1) as an integrase inhibitor [
34,
35,
36,
37,
38,
39]. In addition, the hit compound ZINC000014944915 has been reported against influenza virus [
40]. Compound ZINC000013473324 is an antiviral agent with reported activity against human rhinovirus A protease [
41]. Finally, compound ZINC000001651128 is a curcumin with various activity especially reported as an inhibitor of the human immunodeficiency virus-type1 (HIV-1) integrase [
42]. These findings proves that our potential hits discoveries are indeed antiviral agents and are potentially active against the SARS-CoV-2 RdRp. The stabilities of the hit compounds were further studied via MD simulation.
3.3. Molecular Dynamics Simulation
MD simulation is an appealing method for real-time studies of the conformational stability and dynamics of a protein upon ligand binding. Ten-nanosecond simulation studies were performed on 11 selected systems (complexes including compounds ZINC000014944915, ZINC000027556215, ZINC000013556344, ZINC000003589958, ZINC000003833965, ZINC000001642252, ZINC000028525778, ZINC000027557701, ZINC000013781295, ZINC000001651128 and ZINC000013473324).
3.3.1. RMSD Analysis
By measuring the variance from the original configuration, the RMSD was calculated as part of the MD simulations to estimate the overall stability of the protein complex upon ligand binding. Ten of the 11 MD simulation systems were remarkably stable. All systems except ZINC000027557701 exhibited RMSD values of around 3 Å with average RMSD values of 1.81 ± 0.305 Å to 2.141 ± 0.425 Å (
Figure S1 and
Table S2;
Supplementary Materials). Notably, slight fluctuation was observed in the Cα backbone around 10,300 ps. However, the Cα backbone was stabilized for the remainder of the simulation, indicating system convergence. These results showed stable internal motion in all systems, except that containing ZINC000027557701.
3.3.2. RMSF Analysis
RMSF values of the protein residues were obtained via least-squares fitting to the initial structural configuration as the control frame over a 10-ns trajectory. The RMSF values of the RdRp protein residues with no ligand bound were calculated and compared to those after compound binding (
Figure S2;
Supplementary Materials). After binding the compound to the RdRp active site, the RMSF values of the residues revealed slight fluctuation of the backbone residues. The loop region (
Figure 4 fluctuated slightly across all systems, while active residue regions remained unchanged during the simulation process. The results show that interaction with all 11 hits stabilized the target protein. The binding of all selected hits into the active site reduced variations in the loop in terms of only protein structural fluctuations. However, ZINC000027557701 binding in the active site resulted in further fluctuations in the protein structure, which were primarily observed in the 30th and 33rd helix (
Figure 4). Overall, when the selected ligands bound to the target protein, the main protein backbone conformation remained unchanged. The active binding domain was estimated to include the 29th and 33rd helix regions and the beta-turn between the two F strands, as shown in
Figure 4. Significant sharp peaks were observed for residues 630 to 640 and 686 to 694, which are close to these structures.
3.3.3. Radius of Gyration
The radius of gyration provides information about protein structural folding and rearrangement after ligand binding. Thus, the R
oG was studied to evaluate the system compactness over time. More protein unfolding (less compactness) is represented by higher R
oG values, while more protein folding (more compactness) is associated with lower R
oG values and greater structural stability. All systems yielded estimated average R
oG values between 31.0 and 31.4 Å (
Figure S3; Supplementary Materials). The average gyration scores of all selected systems were estimated and are tabulated in
Table S2 (Supplementary Materials). The data show that, during simulation, all systems were compact (except ZINC000027557701, which produced a higher R
oG value), suggesting that the systems converged well.
3.3.4. Hydrogen Bond Stability
The hydrogen bond stability was determined to further validate the stability of each complex. All possible hydrogen donors and acceptors in the active site domain of the RdRp protein were estimated during the hydrogen bond stability analyses. The angles between the acceptor and donor were set at 30°, and the geometric criteria were maintained ≤3.5 Å. The hydrogen bonds formed between the compounds and the active residues within the RdRp protein were properly maintained throughout the MD simulations. The average hydrogen bonding interactions of all selected compounds are shown in
Table S2 (Supplementary Materials). Compounds ZINC000003589958 and ZINC000013556344 showed more favorable average hydrogen bonding interactions than those of the other compounds. Additionally, it can be observed that all selected compounds formed direct hydrogen bonds with the catalytic residue Asp760 at zero time and were undirected for a limited period of simulation. Compounds ZINC000003589958 and ZINC000013556344 formed direct hydrogen bonds with Asp686 and Arg550 during the simulation process (see
Table S2 and
Figure S4; Supplementary Materials).
3.4. ADMET Study
To support our effort to understand the pharmacokinetic properties and toxicity effects of the newly identified hits, we studied the ADMET properties of the selected candidates. The selected compounds were subjected to drug-like property analyses according to Veber rules [
43] and the Lipinski rule of five [
44] (
Table S3;
Supplementary Materials).
Discovery Studio 2016 was utilized to calculate the ADMET parameters, and the results are given in
Table S4 (Supplementary Materials) [
20]. Furthermore, the confidence levels of the predictions provided by the BBB and the HIA models were determined by constructing the ADMET plot. The calculated AlogP98 is plotted against the PSA_2D for the selected candidates in
Figure S5 (Supplementary Materials). Two analogs are represented in this plot, namely, the 95% and 99% confidence ellipses, which correspond to the HIA and BBB models, respectively. There is an inverse relationship between the PSA value of a given compound and its human intestinal absorption value, which also extends to the cell wall permeability [
23]. Moreover, since the value of AlogP98 is a ratio that indicates lipophilicity, it can be used to calculate the hydrophilicity and hydrophobicity of a compound. As a result, hydrogen bonding characteristics can be obtained by calculating the PSA and AlogP98 [
21]. The regions of chemical space that contain well-absorbed compounds (≥90%) are indicated by the 95% confidence ellipses, and the regions that contain compounds with improved absorption through the cell membrane are represented by the 99% confidence ellipses. In general, a compound with good cell permeability should have PSA and AlogP98 values <140 Å
2 and <5, respectively [
21].
The selected compounds were subjected to ADMET studies. Compounds ZINC000001642252, ZINC000003833965, ZINC000013473324, ZINC000013781295, and ZINC000014944915 showed good human intestinal absorption (absorption level 0), and compounds ZINC000001651128 and ZINC000028525778 showed moderate absorption (absorption level 1). Compounds ZINC000013556344 and ZINC000027557701 showed low absorption (absorption level 2), and the remaining compounds showed very low absorption (absorption level 3). Upon investigating aqueous solubility, it was found that compounds ZINC000013556344, ZINC000027556215, and ZINC000027557701 showed low aqueous solubility (solubility level 2), while compounds ZINC000001642252, ZINC000001651128, ZINC000003589958, ZINC000003833965, ZINC000013473324, ZINC000013781295, and ZINC000028525778 showed good aqueous solubility (solubility level 3). Compound ZINC000014944915 showed optimal solubility (solubility level 4). Compounds ZINC000014944915 and ZINC000028525778 showed probable hepatotoxicity, while all other compounds showed non-hepatotoxic properties. Compounds ZINC000001642252, ZINC000001651128, ZINC000013473324, ZINC000013556344, ZINC000027557701, and ZINC000028525778 showed plasma protein binding, while all other compounds did not exhibit plasma protein binding properties. All selected compounds except ZINC000003589958, ZINC000013556344, and ZINC000027556215 fell inside the ADME model ellipse filter, suggesting good intestinal absorption and BBB penetration ability.
4. Conclusions
To discover potential SARS-CoV-2 RdRp inhibitors, we performed computational drug discovery techniques, including database optimization, virtual screening, MD simulations, and ADMET calculations and estimations. The antiviral ZINC database was utilized for database preparation and screening. The SARS-CoV-2 RdRp was retrieved and optimized for virtual screening and molecular docking analyses. Approximately 13,840 ZINC antiviral compounds were used for covalent docking virtual screening. In total, 241 of these compounds were shown to interact with the RdRp domain covalently, and subsequent filtration based on docking scores yielded 85 compounds. Detailed molecular docking interaction analyses based on the interactions of the hits with previously reported important residues within the RdRp domain identified 11 compounds. These top hits were utilized for MD simulations, including RMSD, RMSF, Rog, and hydrogen bonding stabilization studies. ADMET studies were also carried out on the top hits.
In the virtual screening analysis, the top five compounds, ZINC000014944915, ZINC000027556215, ZINC000013556344, ZINC000003833965, and ZINC000001642252, displayed the highest binding affinity scores of −20.37, −20.19, −17.91, −17.02 and −16.86 kcal/mol, respectively. In the RMSD studies, all hit compounds showed average RMSD values of 1.81 ± 0.305 Å to 2.141 ± 0.425 Å, except the ZINC000027557701 system. The RMSF studies showed that interaction with the hits stabilized the target protein, except in the case of ZINC000027557701, which caused fluctuations in the protein structure. RoG values revealed that all studied systems were compact, except the system containing ZINC000027557701. The hydrogen bond stabilization studies showed that all compounds interacted well with the active residues within the binding domain of RdRp. The ADMET analyses showed that no hits were hepatotoxic except ZINC000014944915 and ZINC000028525778. All compounds displayed good solubility, with no inhibition of the CYP2D6 enzyme. Moreover, all hit molecules showed good to moderate absorption levels, except for ZINC000003589958 and ZINC000027556215, which showed low absorption levels. Lastly, six hits—ZINC000001642252, ZINC000001651128, ZINC000013473324, ZINC000013556344, ZINC000027557701, and ZINC000028525778—showed plasma protein binding, while the remaining hits did not.
In conclusion, the hit molecules ZINC000003833965, ZINC000001642252, ZINC000013781295, and ZINC000013473324 are potential RdRp inhibitors that display better properties compared to the other hits. These four hits can be further studied and optimized to pave the way for developing better anti-COVID-19 agents. Further experimental studies are needed to confirm these findings.
Supplementary Materials
The following are available online at
https://www.mdpi.com/article/10.3390/cryst11050471/s1, Figure S1: RMSD of the hit compounds and the protein calculated as a function of time over a 10 ns run; Figure S2: RMSF of the hit compounds and amino acid residues calculated as a function of time over a 10 ns run; Figure S3: Radius of gyration of the protein and the 11 compounds calculated as a function of time over a 10 ns run; Figure S4: Hydrogen bond stabilization of the 11 compounds over 10 ns production run; Figure S5: ADMET plot for the selected hits; Table S1: Molecular docking interaction between RdRp and selected compounds; Table S2: The average of gyration scores and RMSD as well as the number of H-bonds for selected compounds; Table S3: Physicochemical properties of the hit compounds based on Lipinski’s rule-of-five; Table S4: ADMET values of selected hit compounds.
Author Contributions
Formal analysis, A.H.B., A.R.H. and H.M.A.; investigation, M.Y.G., A.H.B. and H.M.A.; methodology, M.Y.G., A.H.B. and A.A.A.; project administration, A.A.A.; software, A.H.B. and A.R.H.; validation, A.R.H. and A.A.A.; visualization, A.R.H.; writing—original draft, M.Y.G., H.M.A. and A.A.A.; writing—review & editing, M.Y.G., A.R.H. and A.A.A. All authors have read and agreed to the published version of the manuscript.
Funding
Funding was obtained via the Institute of Research and Consulting Studies at King Khalid University and The Deanship of Scientific Research at King Saud University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available within the article and in the supplementary materials.
Acknowledgments
The authors are thankful to the Institute of Research and Consulting Studies at King Khalid University of supporting this research through grant number 12-64-S-2020. The authors also extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group No. RG-1440-052.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Zhang, L. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.F.-W.; Yuan, S.; Kok, K.-H.; To, K.K.-W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Wang, X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Ziebuhr, J. The Coronavirus Replicase. Curr. Top. Microbiol. Immunol. 2005, 287, 57–94. [Google Scholar]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef] [Green Version]
- Elfiky, A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020, 248, 117477. [Google Scholar] [CrossRef]
- Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Götte, M. Mechanism of Inhibition of Ebola Virus RNA-Dependent RNA Polymerase by Remdesivir. Viruses 2019, 11, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Crystal Structure of the 2019-nCoV Main Protease Complexed with Boceprevir. Available online: http://www.rcsb.org/structure/7BRP (accessed on 15 April 2020).
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins: Struct. Funct. Bioinform. 2008, 75, 187–205. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment (MOE). 2015.10; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910 Montreal, QC, Canada, H3A 2R7. 2015. [Google Scholar]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Shi, J.-H.; Pan, D.-Q.; Jiang, M.; Liu, T.-T.; Wang, Q. Binding interaction of ramipril with bovine serum albumin (BSA): Insights from multi-spectroscopy and molecular docking methods. J. Photochem. Photobiol. B Biol. 2016, 164, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Discovery Studio Modeling Environment; Release 2017; DassaultSystèmes: San Diego, CA, USA, 2016; Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 1 September 2016).
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Merz, K.M. Prediction of aqueous solubility of a diverse set of compounds using quantitative structure−property relationships. J. Med. Chem. 2003, 46, 3572–3580. [Google Scholar] [CrossRef]
- Egan, W.J.; Lauri, G. Prediction of intestinal permeability. Adv. Drug Deliv. Rev. 2002, 54, 273–289. [Google Scholar] [CrossRef]
- Dixon, S.L.; Merz, K.M. One-Dimensional Molecular Representations and Similarity Calculations: Methodology and Validation. J. Med. Chem. 2001, 44, 3795–3809. [Google Scholar] [CrossRef]
- Cheng, A.; Dixon, S.L. In silico models for the prediction of dose-dependent human hepatotoxicity. J. Comput. Mol. Des. 2003, 17, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Ziebuhr, J. The coronavirus replicase. In Coronavirus Replication and Reverse Genetics; Springer: Berlin/Heidelberg, Germany, 2005; pp. 57–94. [Google Scholar]
- Peng, Q.; Peng, R.; Yuan, B.; Zhao, J.; Wang, M.; Wang, X.; Shi, Y. Structural and Biochemical Characterization of the nsp12-nsp7-nsp8 Core Polymerase Complex from SARS-CoV-2. Cell Rep. 2020, 31, 107774. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, K.C.; Gulyaeva, A.; Zevenhoven-Dobbe, J.C.; Janssen, G.M.C.; Ruben, M.; Overkleeft, H.S.; Van Veelen, P.A.; Samborskiy, D.V.; Kravchenko, A.A.; Leontovich, A.M.; et al. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015, 43, 8416–8434. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Song, H.; Wang, H.; Chai, Y.; Su, C.; Qi, J.; Shi, Y.; Gao, G.F. The crystal structure of Zika virus NS 5 reveals conserved drug targets. EMBO J. 2017, 36, 919–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengrung, N.; El Omari, K.; Martin, I.S.; Vreede, F.T.; Cusack, S.; Rambo, R.P.; Vonrhein, C.; Bricogne, G.; Stuart, D.I.; Grimes, J.M.; et al. Crystal structure of the RNA-dependent RNA polymerase from influenza C virus. Nat. Cell Biol. 2015, 527, 114–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy, A.S.; Lima, G.M.A.; Oliveira, K.I.Z.; Torres, N.U.; Maluf, F.V.; Guido, R.V.C.; Oliva, G. Crystal structure of Zika virus NS5 RNA-dependent RNA polymerase. Nat. Commun. 2017, 8, 14764. [Google Scholar] [CrossRef]
- Gong, P.; Peersen, O.B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2010, 107, 22505–22510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Xu, H.E. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- King, P.J.; Ma, G.; Miao, W.; Jia, Q.; McDougall, B.R.; Reinecke, M.G.; Cornell, C.; Kuan, J.; Kim, T.R.; Robinson, W.E. Structure−Activity Relationships: Analogues of the Dicaffeoylquinic and Dicaffeoyltartaric Acids as Potent Inhibitors of Human Immunodeficiency Virus Type 1 Integrase and Replication†. J. Med. Chem. 1999, 42, 497–509. [Google Scholar] [CrossRef]
- Mustata, G.I.; Brigo, A.; Briggs, J.M. HIV-1 integrase pharmacophore model derived from diverse classes of inhibitors. Bioorganic Med. Chem. Lett. 2004, 14, 1447–1454. [Google Scholar] [CrossRef]
- Buolamwini, J.K.; Assefa, H. CoMFA and CoMSIA 3D QSAR and Docking Studies on Conformationally-Restrained Cinnamoyl HIV-1 Integrase Inhibitors: Exploration of a Binding Mode at the Active Site. J. Med. Chem. 2002, 45, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.R.; Fesen, M.R.; Mazumder, A.; Wang, J.; Carothers, A.M.; Grunberger, D.; Driscoll, J.; Kohn, K.; Pommier, Y. Hydroxylated aromatic inhibitors of HIV-1 integrase. J Med. Chem 1995, 38, 4171–4178. [Google Scholar] [CrossRef] [PubMed]
- Bailly, F.; Queffelec, C.; Mbemba, G.; Mouscadet, J.-F.; Cotelle, P. Synthesis and HIV-1 integrase inhibitory activities of caffeic acid dimers derived from Salvia officinalis. Bioorganic Med. Chem. Lett. 2005, 15, 5053–5056. [Google Scholar] [CrossRef] [PubMed]
- Artico, M.; Di Santo, R.; Costi, R.; Novellino, E.; Greco, G.; Massa, S.; Tramontano, E.; Marongiu, M.E.; De Montis, A.; La Colla, P. Geometrically and Conformationally Restrained Cinnamoyl Compounds as Inhibitors of HIV-1 Integrase: Synthesis, Biological Evaluation, and Molecular Modeling. J. Med. Chem. 1998, 41, 3948–3960. [Google Scholar] [CrossRef] [PubMed]
- Lew, W.; Wu, H.; Mendel, D.B.; Escarpe, P.A.; Chen, X.; Laver, W.; Graves, B.J.; Kim, C.U. A new series of C3-aza carbocyclic influenza neuraminidase inhibitors: Synthesis and inhibitory activity. Bioorganic Med. Chem. Lett. 1998, 8, 3321–3324. [Google Scholar] [CrossRef]
- Johnson, T.O.; Hua, Y.; Luu, H.T.; Brown, E.L.; Chan, F.; Chu, S.S.; Dragovich, P.S.; Eastman, B.W.; Ferre, R.A.; Fuhrman, S.A.; et al. Structure-Based Design of a Parallel Synthetic Array Directed Toward the Discovery of Irreversible Inhibitors of Human Rhinovirus 3C Protease. J. Med. Chem. 2002, 45, 2016–2023. [Google Scholar] [CrossRef]
- Mazumder, A.; Neamati, N.; Sunder, S.; Schulz, J.; Pertz, H.; Eich, E.; Pommier, Y. Curcumin Analogs with Altered Potencies against HIV-1 Integrase as Probes for Biochemical Mechanisms of Drug Action. J. Med. Chem. 1997, 40, 3057–3063. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










