1. Introduction
Electronic correlations together with a complex interplay between charge, spin, and lattice degrees of freedom in transition metal oxides with perovskite structure result in interesting physical phenomena and promising functional properties, such as colossal magnetoresistance (CMR) in mixed-valence manganites [
1,
2] and high-T
C superconductivity in cuprates [
3,
4]. The possibility of controlling these intriguing phenomena and using them in potential device applications have motivated long-term research in perovskite oxides with the general formula ABO
3. The A and B positions can be occupied by rare earth (La, Pr, Nd, etc.) and transition metal (Mn, Co, Ni, etc.) cations, respectively, having different oxidation states (valences) and cation sizes. Moreover, the perovskite structure enables systematic chemical doping and substitution on both A- (A
1−xA’
xBO
3) and B-sites (AB
xB’
1−xO
3), with A’ as di-(Ca, Sr, Ba), tri-(La, Pr, Nd), or mono-valent (Li, Na) cations, and BB’ as transition metal cations, e.g., Ni, Mn, Co, etc. In addition to the random distribution of cations between the A- and B-positions, such as that occurring in an alloy, a cation ordering within the A- or B-sites or even a simultaneous ordering within A- and B-sites in the perovskite structure can be achieved [
5,
6,
7,
8,
9].
Cation ordering can play a crucial role in the above-mentioned interplay of charge, spin, and lattice degrees of freedoms, resulting in specific electrical and magnetic properties, which cannot be realized in a cation-disordered material. Particularly interesting are the so-called double perovskites (DP) with the sum formula A
2BB’O
6, where B-sites are occupied in an ordered manner by two different cations with a B/B’ ratio of 1:1. The degree of the B-site ordering determines the crystal structure and magnetic exchange interactions between the respective magnetic B-cations. Generally, B-site ordering can be controlled by
steric (cation sizes) and
Coulomb interactions (oxidation states) between the chosen B/B’ cations [
7,
8], which can be considered intrinsic conditions. However, the realizations of B-site ordering are strongly influenced by the
growth conditions of a DP material as well as by unavoidable one-dimensional (vacancies and impurities) and two-dimensional (interfaces and grain boundaries)
defects. All these intrinsic and extrinsic material aspects, preventing an ideal 100% B-site ordering, nevertheless allow researchers to control the physical and physico-chemical properties of DP to a large extent. Striking examples of such a control provide: (a) the half-metallic double perovskite Sr
2FeMoO
6 [
10], in which the spin polarization of electrons at the Fermi level as well as magnetism are drastically enhanced by the degree of B-site ordering, and (b) the dielectric Pb
2ScTaO
6, displaying a ferroelectric or relaxor dielectric behavior for the fully ordered or disordered Sc/Ta distribution, respectively [
11]. Great interest in the potential applications of DPs is motivated by observations of pronounced magnetodielectric effects [
12,
13,
14], photo- and electro-catalysis [
15], as well as by very recent photovoltaic studies [
15,
16,
17].
Our motivation to write this short review is based on the conviction that the further development of new routes and techniques for the growth of cation-ordered correlated oxide films could produce an essential breakthrough in both fundamental and applied research. Indeed, the study of correlation effects in condensed matter without chemical disorder could particularly impact the areas of classical and quantum phase transitions, low-dimensional systems, and nanoscale physics. Potential applications would strongly benefit from the development of advanced growth routes that could controllably and reproducibly establish a high degree of cation ordering by the fine tuning of processing conditions at the atomic scale.
This review article is organized in such a way that we begin (part 2) with a short discussion of the general aspects and models of cation ordering in bulk DPs. In the third part, we address the B-site ordering and its quantification and manifestations in thin films, prepared by different physical vapor deposition (PVD) techniques such as pulsed laser deposition (PLD), sputtering, molecular beam epitaxy (MBE), and chemical routes such as metalorganic aerosol deposition (MAD) and polymer-assisted deposition (PAD). At this point, the discussion is focused on the influence of specific growth conditions inherent to different deposition techniques as well as on the epitaxial strain actuated by the different single crystalline substrates. We have mainly limited ourselves to the La2CoMnO6 (LCMO) and La2NiMnO6 (LNMO) films as the most studied magnetic DP materials. Finally, the recent results on the realization of B-site ordering at interfaces in perovskite superlattices, as well as the application of the layer-by-layer growth, will be presented and discussed.
2. General Aspects of B-Site Cation Ordering in Bulk Double Perovskites
If the different B cations are randomly distributed in the crystal, the DP material is considered to be disordered and the notation AB
0.5B’
0.5O
3 is used, while for alternatively occupied B-sites, DP is viewed to be ordered and the notation is A
2BB’O
6. The B-site arrangements of the ordered A
2BB’O
6 DP (visualized by the “Vesta” program [
18]), can be set in three ways [
7,
8], as shown in
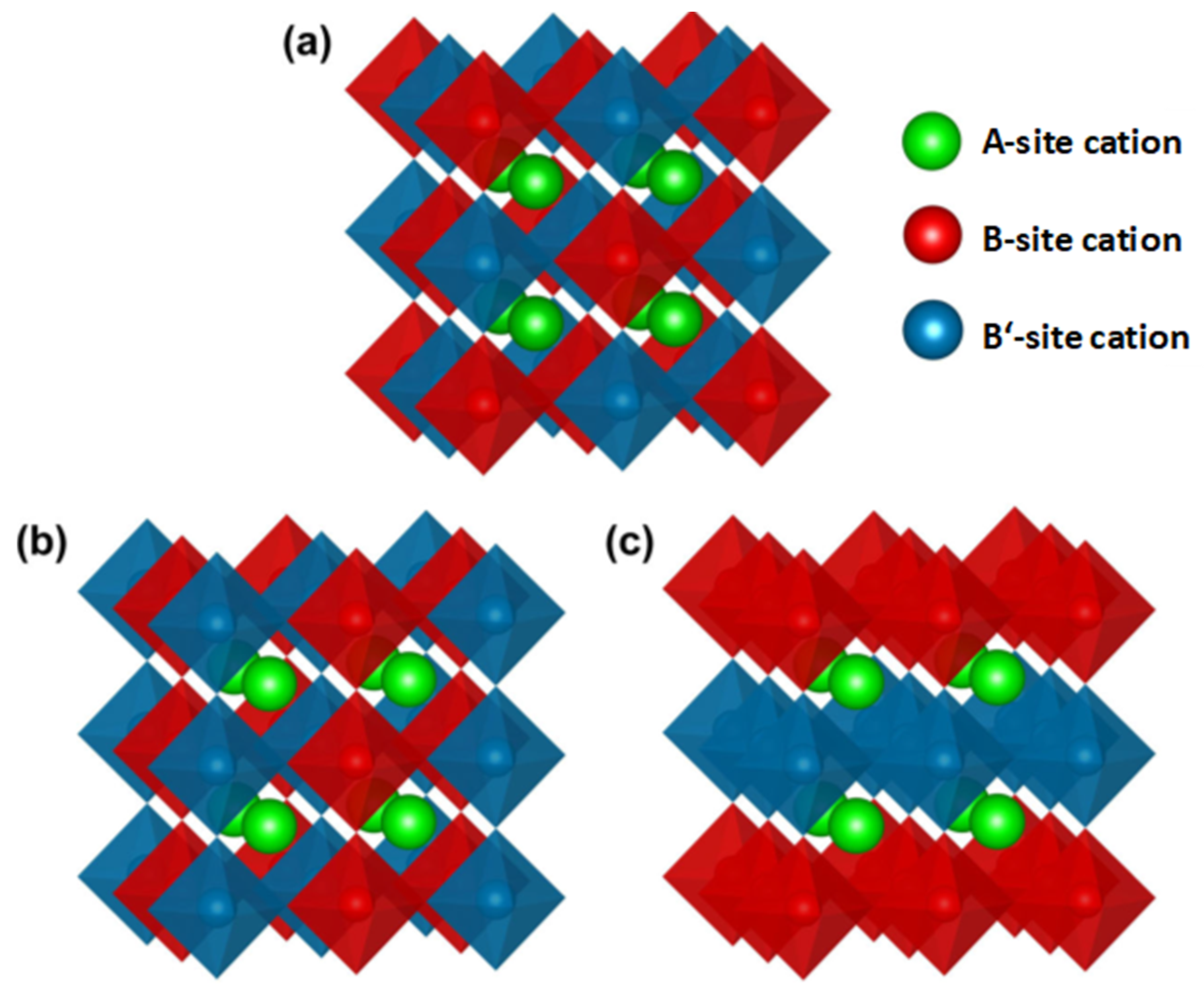
Figure 1a the NaCl type of ordering with B and B’ occupations alternating in all three spatial directions;
Figure 1b the columnar order with alternating B/B’ occupations in two spatial directions; and
Figure 1c the B/B’ layered type of ordering, with alternating planes occupied by B and B’ cations. Since in most cases the B and B’ cations possess different valences or oxidation states in the ordered case, the NaCl type of ordering is energetically favorable from the electrostatic point of view, as B and B’ cations are spatially separated from each other [
7,
8]. This is the reason why the NaCl type of ordering is the most observed type of B-site ordering.
Similar to a simple perovskite oxide ABO
3, the crystal structure of the B-site ordered DP is influenced by A- and B-site cation radii, as described by the tolerance factor t = (r
A + r
O)/√2(r
B + r
O) and the BO
6 octahedral tilt [
8,
19,
20]. In the ideal case of fully matched A- and B-cations, t = 1 and the undistorted
Fm3m cubic structure would be realized. In the most practically relevant cases, the tolerance factors are smaller (t < 1) and the crystal symmetry is reduced to the tetragonal
I4/m or monoclinic
P21/n space group. In contrast, the disordered DPs often crystallize in an orthorhombic
Pbnm structure [
5,
8,
21,
22,
23,
24,
25]. Assuming an ideal NaCl arrangement of B/B’ cations (t = 1), an enlarged unit cell (u.c.) composed of two cubic perovskite u.c. with B and B’ cations in each one, can be considered a
DP = 2a
p and given the name “double perovskite”. This issue becomes particularly clear when considering crystallographic planes with only the same cations, i.e., planes with B- or B’-cations, oriented perpendicularly to the [111]-axis within the NaCl structure of ordering (see
Figure 2). One can view this as a crystal superstructure, composed of the ABO
3 and AB’O
3 perovskite atomic layers, [
8,
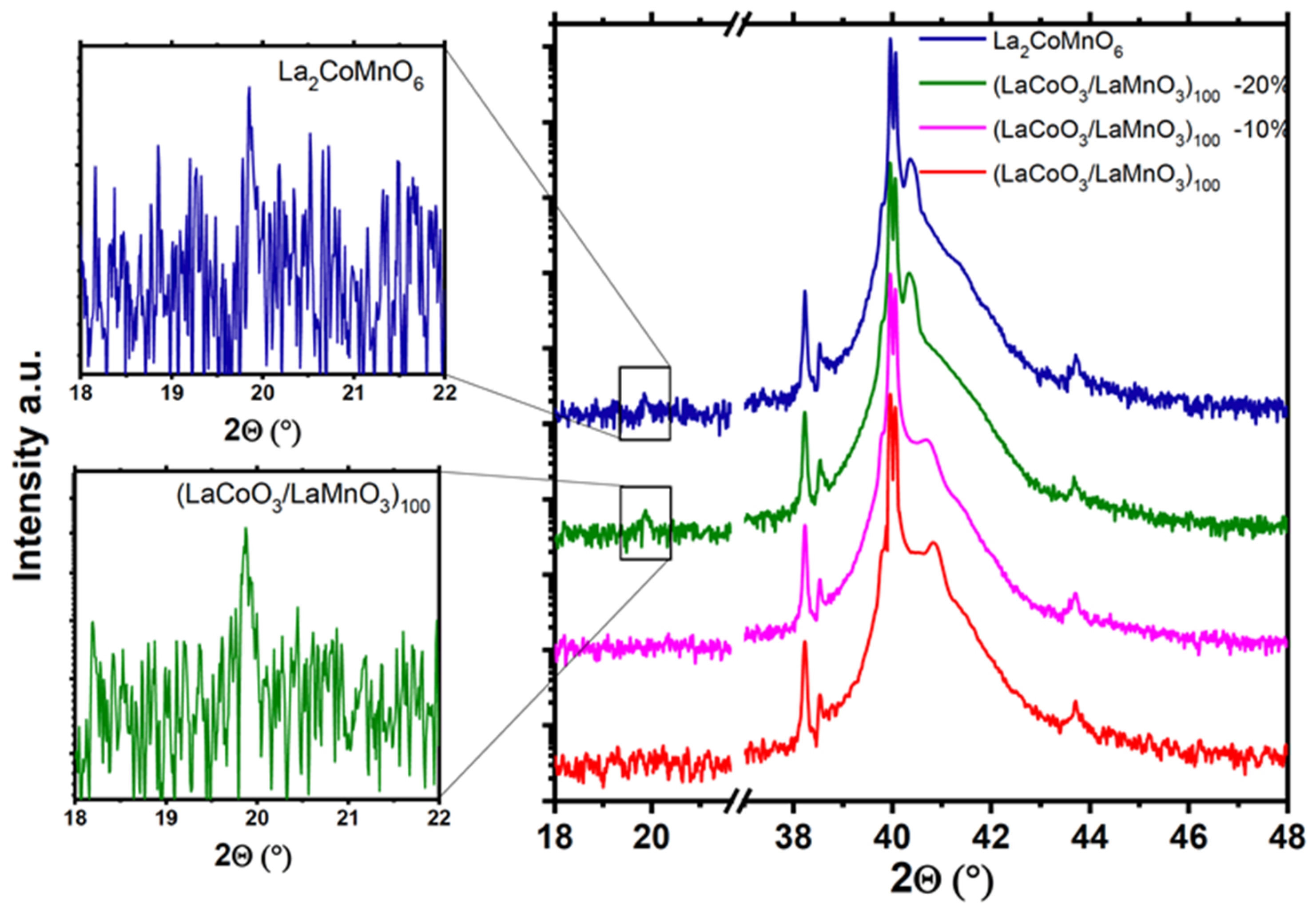
26] which appears due to the doubling of the unit cell in the ordered DP along the [111] direction. Such a superstructure could be resolved in X-ray diffraction (XRD) measurements as an additional superstructure peak (1/2 1/2 1/2), which can be assigned to the doubled u.c. of the ordered DP.
As already mentioned, the physical properties are strongly influenced by the degree of B-site ordering, which as well depends on various driving forces during crystal growth. Considering the NaCl type of ordering, the electrostatics, and thus the difference in valences of the B cations ∆Z
B = |Z
B − Z
B’|, play an important role. Specifically, the larger the B/B’ valence mismatch, the higher the degree of order since this maximizes the distance between the B and B’ cations viewed as effective ionic charges Z
B and Z
B’. Empirical studies show that DPs with a valence difference of ∆Z
B > 2 are usually ordered, and those with a difference of ∆Z
B < 2 are disordered. For ∆Z
B = 2, both ordered and disordered cases are possible [
7,
8]. The second driving factor of B-site ordering is the cation size (radii) mismatch ∆R
B = |R
B − R
B’|: the larger the ∆R
B, the larger the local lattice strain, which thus drives B-site ordering. Interestingly, for an LCMO DP with Co
2+ and Mn
4+ cations in the fully ordered state, the valence and site mismatches are coupled with each other in ∆R = |R
Co2+ − R
Mn4+| = 21 pm, which is considerably larger than the size mismatch between Co
3+ and Mn
3+ cations in ∆R = |R
Co3+ − R
Mn3+| = 3 pm. A similar issue can be observed in the Ni/Mn ordering as ∆R = |R
Ni2+ − R
Mn4+| = 16 pm >> ∆R = |R
Ni3+ − R
Mn3+| = 4 pm. The values of cation radii were taken from ref. [
27]. From the energetic point of view, the gain in the potential electrostatic (Coulomb) energy will be obtained when both Co
2+ and Mn
4+ are separated from each other. But this has to be compensated by the increased elastic energy of the lattice due to local strains as the size mismatch between cations in the ordered phase (Co
2+ and Mn
4+) is much larger than the size mismatch in a fully disordered state (Co
3+ and Mn
3+). It looks probable that the NaCl type of ordering is favorable for the compensation of both charge and size mismatches.
In addition to the two mentioned fundamental driving forces determined by the choice of cation types, the B-site ordering can also be influenced by deposition conditions (growth temperature and growth rate), and for thin films, by the choice of substrate and thus, the epitaxial strain [
5,
8,
26]. The choice of the A-site cation can also affect a DP material as the oxidation state of the A-site cation influences the average valence of the B/B’ cations. For an A
2+ cation (e.g., Ca, Sr, Ba), the average valence of the B cations is four and the ordering can theoretically be achieved for the following valence combinations: B
4+/B’
4+, B
3+/B’
5+, B
2+/B’
6+, and finally, B
1+/B’
7+. On the other hand, an A
3+ cation (La, Pr, Nd etc.) requires that BB’ cations have an average valence of 3+, making possible the following options: B
3+/B’
3+, B
2+/B’
4+, and B
1+/B’
5+ [
8]. Which valence distribution finally occurs depends not only on the B-order but also on the choice of elements for the cations as each element can have its preferred individual valence state according to the applied growth conditions, e.g., oxygen partial pressure.
To quantify the B-site ordering, a long-range degree of ordering, s = 2g
B − 1, is introduced; here, g
B denotes the occupation of correct lattice sites by the B and B’ cations [
8]. The completely ordered DP possesses a value of s = 1, which means that all sites are fully occupied by the correct cations and the so-called anti-site disorder, AS = 0. A completely disordered DP will have a value of s = 0. In between these two extreme situations, DPs are considered to be partially ordered, with a degree of ordering at 0 < s < 1. Deviations from the fully ordered phase, otherwise known as lattice misoccupations, occur in the form of point defects (PD) with an anti-site disorder, in which the B and B’ cations as well as the antiphase boundaries (APB) are exchanged, separating two ordered domains with opposite B and B’ occupations. PDs and APBs have different effects on the cation ordering in a DP material. In particular, the point defects destroy the short-range B-site ordering, yielding an occupation error and reducing the s-value to some extent. In contrast, the APBs can cause a drastic reduction in the long-range B-site order, yielding very small s~0 values. In the literature, the intensity ratio of the Bragg structural XRD peak, with (hkl) = (111), and of the superstructure peak (1/2 1/2 1/2) is often used to estimate the order by means of the parameter S = I(½ ½ ½)/I(111). However, this estimation of the local B-site order should be treated with caution; despite a high degree of local B-site ordering, the superstructure peak can be totally extinguished due to the presence of the APB domains with opposite orientations lying antiparallel to the scattering vector, thus giving a small total
s-value. In bulk DP materials, B-site ordering can also be quantified by the ratio [I(½ ½ ½)/I(200)]
obs./[I(111)/I(200)]
theo. [
11]; however, this cannot be used for thin epitaxial films because not all reflexes can be simultaneously measured in an XRD pattern.
It is therefore essential, especially for thin film studies, to include additional measurements and experimental data to determine the B-site ordering. These can be magnetization data taken for magnetic DP materials as well as Raman spectra, which were found to be informative as ordered (O) and disordered (D) DP materials crystalized in different crystal structures. With regard to magnetism, in a fully ordered LCMO DP, the superexchange interaction between the Co
2+ and Mn
4+ ions according to the second Goodenough–Kanamori–Anderson rule [
6,
18,
28] results in a maximum value of saturation magnetization M
theo = 6 μ
B/f.u. This value can be used to estimate the “magnetic” degree of order as s
mag = M
meas.(T = 4.2 K)/M
theo. Moreover, the Curie temperature of an ordered DP was found to be significantly higher than that of the disordered variant, i.e., for LCMO T
C(O)~230 K >> T
C(D)~130 K [
6,
18,
28]. Raman spectroscopy is a powerful nondestructive spectroscopic technique, which allows the direct structural characterization of a DP material [
21,
22,
23,
24,
25]. By measuring polarization-dependent Raman spectra, one can distinguish between the disordered (orthorhombic,
Pbnm) and ordered (monoclinic,
P21/n) variants as these structures possess different Raman selection rules. In addition, according to Truong et al. [
25], even the admixture of ordered and disordered phases can be detected by Raman spectroscopy. However, these macroscopic techniques do not address the degree of cation ordering directly; further assumptions and knowledge of magnetic interactions and the structural determination of the phonon spectra are still needed. Finally, a transmission electron microscope (TEM) utilized for chemical mapping by means of atomically resolved electron energy loss spectroscopy (EELS) can be used to directly visualize the local B-site ordering on the atomic scale. However, this is less informative than APBs.
3. Cation Ordering in Thin Films
Nevertheless, the growth of B-site cation-ordered correlated oxide thin films is known to be a challenging task. As already mentioned, it requires growth conditions—e.g., substrate temperature, growth rate, partial pressure of oxygen—to be compatible with those favoring the cation ordering. As mentioned above, the driving forces for cation ordering in bulk (single crystals) oxides are charge and size mismatches between the involved B-site cations; Co
2+ and Mn
4+ as well as the NaCl type of order seem to be favorable for compensation of both charge and size mismatches. Considering the formation free energy of a DP,
E = U − TS (U is the inner energy, T and S are temperature and entropy, respectively), the energy minimum for the ordered phase can be achieved only if the temperatures are high enough as the entropy term for the ordered (O) variant is smaller than that for the disordered (D) one: S
O < S
D. However, at high temperatures, the increased growth kinetics of a DP film would also increase the entropy, hence favoring the disordered state [
8]. Thus, a compromise between the thermodynamics and kinetics has to be found in order to achieve the growth of cation-ordered DP films. It is worth noting that all possible deviations from the stoichiometry, e.g., A/B and B/B’ atomic ratios, and growth-induced defects, e.g., oxygen vacancies and interstitial atoms, would favor the disordered phase by the entropy term. It seems reasonable to assume that the film growth conditions close to the equilibrium, i.e., high temperatures and low degree of supersaturation, together with a fine control of the chemical composition could be favorable for achieving the ordering.
Thin films of DP, including those of LCMO and LNMO, have been prepared by well-elaborated and widely used thin film vacuum deposition techniques, such as PLD, sputtering, and MBE. Particularly noteworthy is the fact that reports on PLD-grown DP films dominate the literature, whereas the MBE is much less presented. In general, the film growth within PLD and sputtering is usually considered a non-equilibrium and kinetically controlled process as the deposition species (atoms, clusters, molecules) possess high kinetic energy and are able to create defects in the growing film. However, although the MBE technique is free from the above-mentioned effects and provides near-to-equilibrium growth conditions, it also operates at low oxygen pressure, pO
2~10
−5 mbar [
29]. This results in oxygen deficiency and weak saturation magnetization in the as-grown MBE-grown films; for example, the LNMO films show only M
sat~1.3 μ
B/f.u. [
29], which increases up to M
sat~3.5 μ
B/f.u. after post-annealing, remaining significantly smaller than the theoretical value M
theo = 5 μ
B/f.u. for a fully ordered LNMO [
23]. Comparing MBE with the PLD and sputtering, the two latter techniques offer higher oxygen pressure, pO
2~0.1 mbar [
30] during deposition. However, the problem of oxygen deficiency and/or preparation-induced defects for PLD-grown films probably still exist in order to obtain strong magnetic properties; for example, in LCMO films [
31] a post-annealing in 500 mbar of oxygen at 700 °C was shown to be a necessary condition. Several reports on the growth of LCMO [
32] and LNMO [
33] films by rf-sputtering under pO
2~0.1–0.5 mbar also pointed out the importance of post-annealing in the improvement of oxygen stoichiometry and magnetism in these films. Truong et al. [
25] reported the influence of growth conditions (temperature/pO
2) on the Ni/Mn ordering, studied by Raman spectroscopy, in the PLD-grown LNMO(111)/STO(111)films. The long-range ordered (O) phase was grown at 800 °C/1 mbar, the disordered (D) phase at 500 °C/0.4 mbar, while the O + D admixture was at 800 °C/0.4 mbar. These results outline the importance of both high growth temperatures and high oxygen pressure to achieve the B-site ordering.
In contrast to the PVD routes, which operate far from equilibrium and at reduced oxygen pressure, chemical routes such as the metalorganic aerosol deposition (MAD) [
34,
35] as well as other solution-based techniques such as PAD [
36] can provide growth conditions close to the equilibrium. Moreover, the MAD growth occurs at a very high partial oxygen pressure, pO
2~200 mbar, usually at ambient air or even in an oxygen-rich atmosphere. All this allows the oxidation of precursors and prevents oxygen deficiency in the grown films. With regard to the MAD growth, the process is based on the heterogeneous pyrolysis reaction of the aerosols of metalorganic precursor solutions on the surface of a heated substrate [
34]. This results in an extremely high-density vapor/liquid phase of precursors and a diffusion-controlled growth rate of v
g~1–5 nm/min due to a high substrate temperature of T
sub~800–950 °C, which ensures a low supersaturation and generally preventing the fast heterogeneous nucleation of 3D clusters. One can compare the MAD growth conditions with those of a liquid phase epitaxy (LPE), yielding a homogeneous and defect-free growth of an oxide film according to the “step-flow” or layer-by-layer 2D growth mode. The advantage is a drastic decrease in the temperature-dependent supersaturation degree, α, and a concomitant increase in the nucleation cluster size, r
S~1/α [
37,
38]. The large size of crystalline domains allows one to reduce the amount and the influence of point defects as well as the possible antiphase boundaries in the grown film. This seems to be important for the control of cation and anion stoichiometry during epitaxy, as we have demonstrated earlier, by the ability to grow the A-site ordered La
3/4Ca
1/4MnO
3 [
39] films, in which the La/Ca order was shown to significantly increase the phase transition temperature. As a result, the MAD growth conditions allow one to obtain epitaxial oxide films and heterostructures, including double perovskites, with high crystalline quality, perfect surface morphology, and optimally high functional properties as was demonstrated earlier for cuprates [
40], manganites [
39], ruthenates [
41], titanites [
34], nanocomposites [
42], and superlattices [
35].
As mentioned above, an important issue for epitaxial DP thin films is the lattice misfit stress. Usually, LCMO and LNMO DP films are grown by PLD and sputtering on single-crystalline functional oxide film substrates with a perovskite structure, e.g., STO, LaAlO
3 (LAO) and (La
0.3Sr
0.7)(Al
0.65Ta
0.35)O
3 (LSAT), with a lattice misfit, ε = 100% ∗ (a
F − a
S)/a
F from −3% to +0.5% [
30,
43]. According to Kleibeuker et al. [
31] the strongest magnetism with M
S(5 K)~6 µ
B/f.u. and T
C~211 K was observed in LCMO film grown on the LSAT(111) substrate, actuating a small compressive in-plane stress. The films grown on STO (small tensile) and LAO (relaxed) substrates display significantly lower M
S(5 K)~3.5–4 µ
B/f.u. and Curie temperatures, especially on the (100) substrates T
C = 113 K (STO) and 125 K (LSAT), pointing out that these films are B-site disordered. However, no structural characterization of the B-site ordering was provided to compare with magnetic data. It is worth noting that in the case of the (100)-oriented substrates, the B-site ordering could be estimated only indirectly from the magnetization data [
14] or Raman spectroscopy [
22,
24].
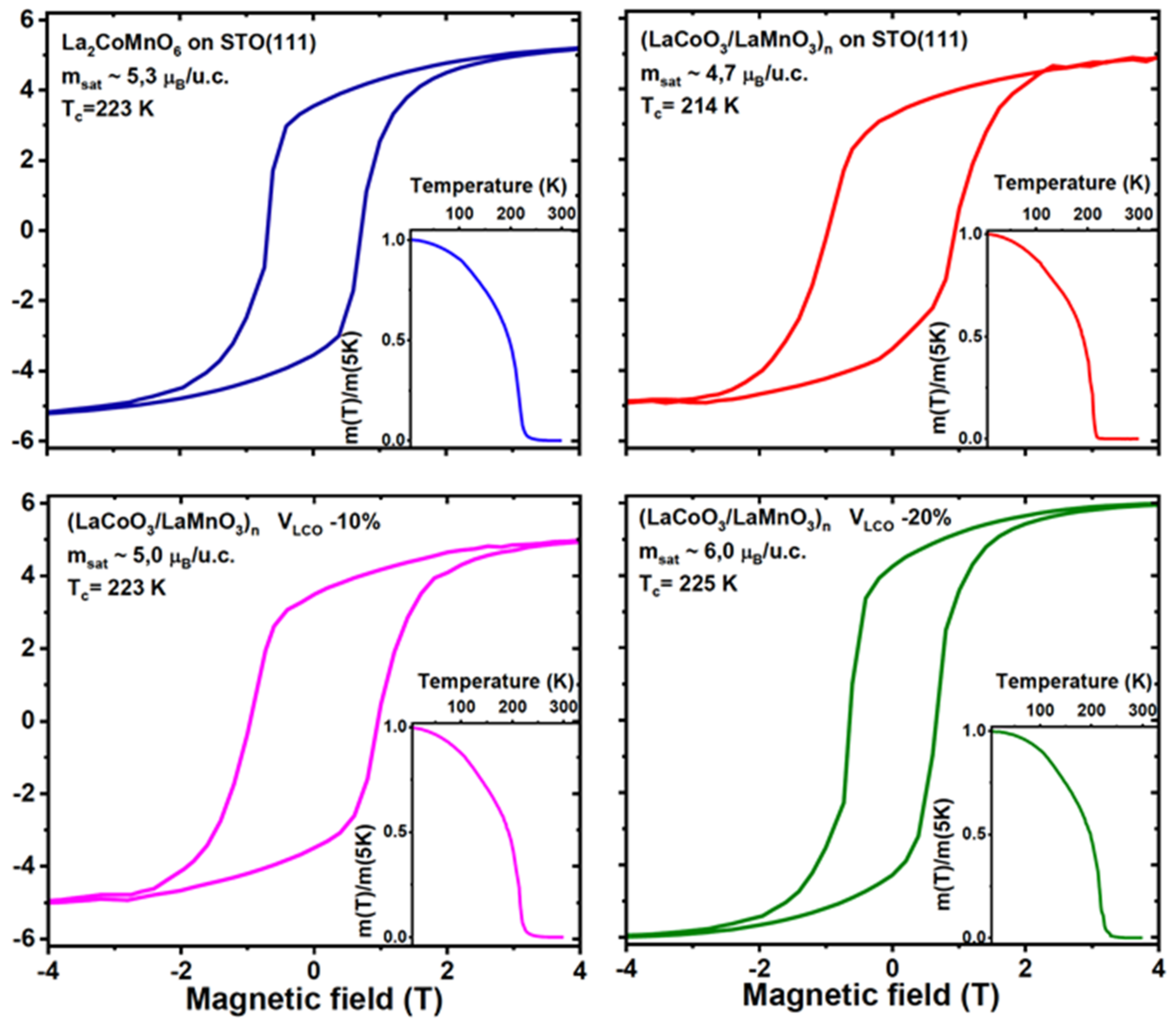
The LCMO films with a high degree of short-range Co/Mn ordering were grown by metalorganic aerosol deposition technique on sapphire [
5] and STO(111) [
6] substrates (see
Figure 3). An atomic force microscopy (AFM) image demonstrates flat surface morphology with the estimated mean-square-roughness, RMS = 0.25 nm. The long-range ordering, quantified by a macroscopic XRD degree of ordering,
s = I(½ ½ ½)/I(111) = 1.1 × 10
−3, was, however, much smaller than the theoretical value,
s = 10
−2, calculated for a fully ordered LCMO, possibly indicating the presence of antiphase boundaries [
6]. The local Co/Mn ordering, obtained by atomically resolved chemical mapping with EELS and shown in
Figure 3, shows the neighboring (111) atomic planes occupied predominantly with Co and Mn ions; however, the full ordering cannot be detected. The valence states of the transition metal cations of Mn
4+ and Co
2+, experimentally determined by TEM and EELS measurements, point to a globally ferromagnetic ground state due to a Co
2+-O-Mn
4+ ferromagnetic super-exchange interaction. High Curie temperatures, T
C = 226 and 228 K, and high saturation magnetization, M
S(4.2 K) = 5.8 and 5.9 µ
B/f.u., were measured for LMCO(111)/STO(111) and LMCO(111)/Al
2O
3(0001) films [
5,
6]. They both indicate a high degree of short-range Co/Mn ordering, s = 0.97, 0.98, estimated as a ratio between the measured saturation magnetization and the maximal theoretical magnetization for the ordered LCMO M
S(theo.) = 6 µ
B/f.c., assuming the ferromagnetically coupled spins of Co
2+ and Mn
4+ ions. Worthy of note is the fact that the disordered LCMO films possess much smaller values of M
S(4.2 K) = 3.5–4.5 µ
B/f.u., as was shown for PLD-grown films earlier [
24,
30].
Comparing the MAD- and PLD-grown thin LCMO films on STO(111) substrates, one can see a clear improvement of ordering in the MAD-grown films. Particularly, in the PLD-grown films, a saturation magnetization of M
S(4.2 K)~4 µ
B/f.c. indicates only a partial ordering with
s~0.67 [
31]. As mentioned in previous studies on PLD-grown LCMO films, non-equilibrium growth conditions result in oxygen vacancies, which are detrimental to B-site ordering [
32]. Even by decreasing deposition temperature and similarly high M
S in LCMO(111)/LSAT(111) grown by PLD [
31], the short-range Co/Mn ordering registered by TEM–EELS imaging is far from being considered perfect. In addition, the EELS spectra taken with low energy resolution do not show fine features inherent in Mn
4+ and Co
2+ ions. It is worth noting that the Co/Mn ordering shown in
Figure 3 for the LCMO(111)/STO(111) film grown by MAD, taken together with clearly defined Co
2+ and Mn
4+ oxidation states in the EELS spectra, allow us to conclude that close-to-equilibrium growth within MAD, accompanied by high oxygen partial pressure, are much more effective for cation ordering than a non-equilibrium growth within PLD and sputtering. Recently, Wang et al. [
36] obtained the Co/Mn ordering in LCMO/STO(100)-grown film by using another near-equilibrium PAD technique. This nontraditional solution-based technique employs metal–polymer solutions as film precursors, which after spin coating onto a substrate and subsequent drying undergo a solid-state reaction at high temperatures of ~900 °C and flow oxygen conditions. The order was confirmed by strong magnetism (T
C = 230 K and M
S~6 μ
B/f.u.) and local TEM–EELS maps, indicating an ordered Co/Mn occupancy of B-sites. Thus, recent PAD and MAD results strongly support the thesis that the near-equilibrium growth conditions, i.e., high temperatures of T
sub~900–950 °C and high pO
2~0.2–1 bar within chemical deposition routes promote the cation ordering. However, even within short-range ordered domains in the MAD-grown films, a complete macroscopic ordering over the whole film thickness is still a challenge due to the presence of defects, such as APBs [
7] as well as twinning domains, with an orientation misfit of ~0.3° observed in LCMO(111)/Al
2O
3(0001) films [
5]. In the latter case, the relaxation of the orientation misfit at the domain boundary by changing the (B,B’)-O bond lengths results in Jahn–Teller distortions and the formation of Mn
3+ and Co
3+ ions.
The DP films grown on (111)-oriented substrates like LCMO/STO(111) [
5,
6,
31] are preferable for studies because both the short- and long-range B-site order can be compared from XRD, TEM–EELS, Raman, and magnetization data. The observed strong dependence of the magnetic properties of a DP material on the crystalline structure and on the B-site ordering can also be interpreted within a magneto-elastic concept, quantified by the so-called spin–phonon coupling constant, λ, which can be directly determined from the temperature-dependent measurements of Raman spectroscopy [
22,
24]. The onset of ferromagnetic ordering at T
C results in a distinct softening of the breathing phonon mode with A
g symmetry, which reflects the increase of a bond length or volume in a ferromagnetic material; meanwhile, as an FM exchange interaction between Co
2+-Mn
4+ spins, described within the fundamental concept of an exchange hole [
44], ions tend to move away from each other. The magnetization-induced change of position of the Raman mode, Δω(T), relative to the anharmonic contribution, ω
anh(T), is described [
22] as follows:
Thus, a spin–phonon interaction can be quantified by the spin–phonon coupling constant, λ, which describes the strength of the magneto-elastic coupling in a DP material and is important for magneto-dielectric applications. In
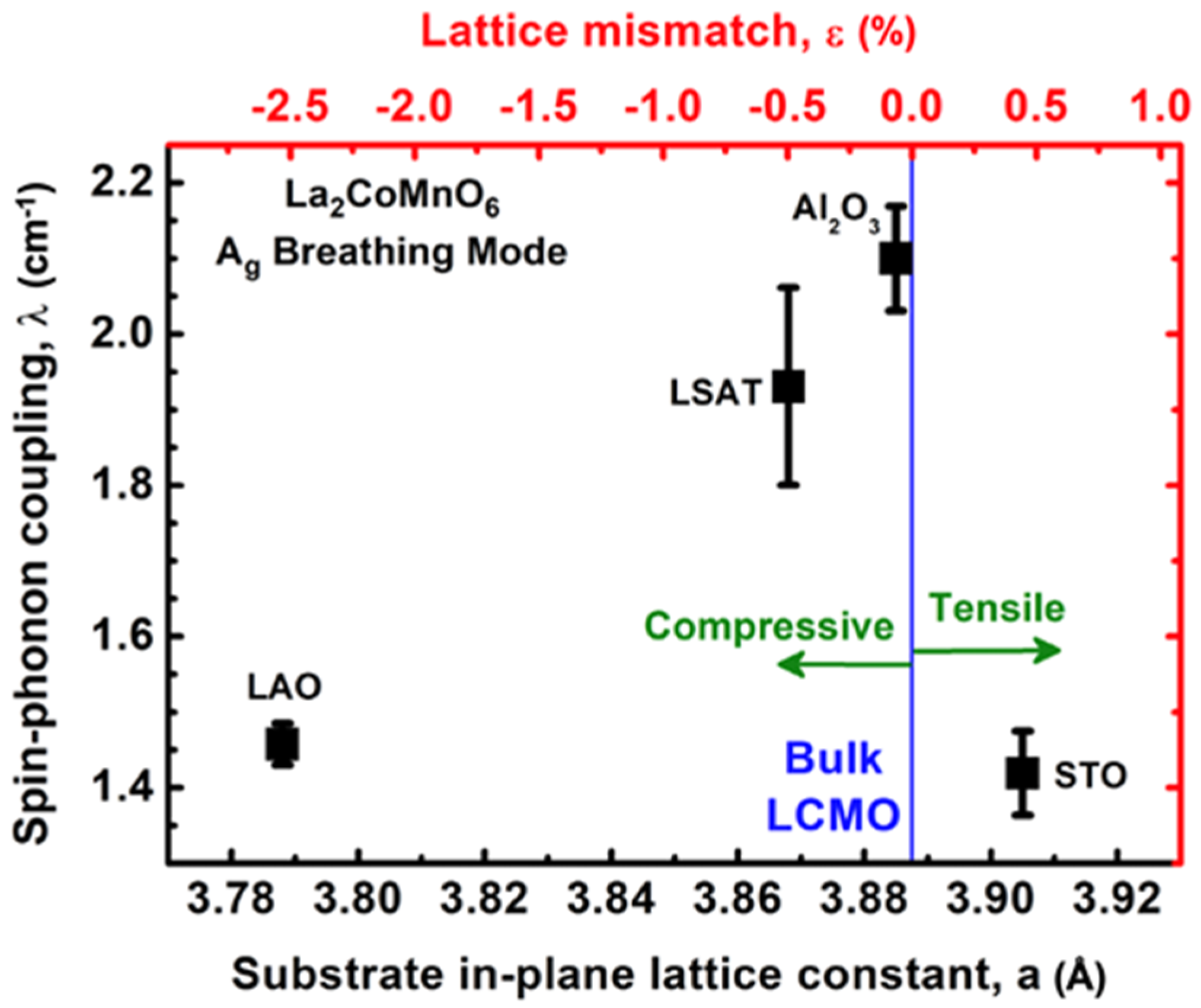
Figure 4, we present the experimentally measured λ values in LCMO films grown on (111)-oriented substrates, actuating different signs and values of epitaxy stress [
5]. One can see that the strongest spin-phonon coupling constant, λ = 2.1 cm
−1, was obtained for the highly ordered LCMO film (s = 0.98) grown on sapphire, which actuated a very small in-plane compressive stress. Moreover, a large lattice mismatch significantly reduced spin–phonon coupling down to λ∼1.5 cm
−1 for LaAlO
3(111) with compressive stress, and to λ∼1.4 cm
−1 for STO(111) with tensile stress. It is worth noting that PLD-grown LCMO films on STO(100) substrates show a value of
λ≈1 cm
−1 [
24].
5. Concluding Remarks
The B-site ordering in double perovskite bulk materials and in thin films, like the mostly studied systems of La2CoMnO6 and La2CoMnO6 is shown to be strongly affected by the mismatch in the B-site cation size and in the valence difference of the B-site cations. Probably, these two factors influence each other and, thus, enable to obtain a high degree of B-site ordering. A comparison between the physical (PLD, sputtering) and chemical (MAD and PAD) growth techniques shows that close to equilibrium growth conditions, easily achieved in the chemical solution-based techniques are favorable for the realization of B-site ordering in thin films. Nevertheless, the commonly used thin film growth methods do not allow a complete B-site order. This circumstance generates a chain of related questions about the ordering in thin films, which we will try to formulate below.
We believe that even with the assumption of the absence of deposition-induced defects and perfect growth conditions, interfaces/surface as unavoidable two-dimensional defects prevent the formation of a fully ordered DP thin film. The next question could be regarding the film thickness at which the influence of interfaces can be minimized. The thickness of the so-called “dead layer” with modified magnetic and structural properties of manganite (La
0.7Sr
0.3MnO
3) films 6–7 u.c.~2–3 nm [
57] can perhaps be taken as a low boundary. This, however, needs to be directly proven by experiments. Another related question is whether a perfect ordering is necessary to obtain the property. Concerning ferromagnetism, the fully ordered DP films possess optimally high T
C and large saturation magnetization, which manifest themselves by macroscopic magnetic measurements, usually at ~80% degree of the B-site ordering. However, the measurement accuracy of the saturation magnetization itself is ~10%, which makes this estimation not very reliable. On the other hand, since the crystal structures of the ordered and disordered phases are different, the admixture films (O + D) can be viewed as multiphase films, which can thus be inclined to degradation. The further question along this line is on how different techniques used to quantify the degree of ordering in thin films, i.e., XRD, Raman, magnetization, and TEM–EELS, can be related to each other? In other words, the degrees of long- and short-range ordering, obtained by XRD and TEM, respectively, do not necessarily correlate with each other. One needs techniques and simulation methods to evaluate the ordering on the mesosocopic scale, comparable with the film thickness, d~20–50 nm. This gap definitely exists as TEM–EELS mapping provides reliable images on a very short (atomic) scale. We believe that an interesting attempt to fill this gap was reported by Spurgeon et al. [
29], as they succeeded in combining the ordering, viewed by TEM–EELS, at the atomic scale of 10–20 nm.
The superlattice approach to the B-site ordering within MAD, e.g., growth of LNO/LMO SLs, was shown to result in an emergent double perovskite phase at the LMO/LNO interfaces. This points out the importance of charge transfer at the interface, likely driven by the differences in the electronegativity between Ni and Mn cations in the realization of ordering. Moreover, the obtained emergent double perovskite LNMO phase in the LMO/SMO SLs is essentially two-dimensional, and likely possesses some unusual magneto-elastic properties deserving further study. Further promising avenues of research would be to apply the SL approach for the growth of DP materials, which do not show B-site ordering in bulk form. We have obtained preliminary results on that problem by comparing the SL and SoA MAD growth of Sr
2RuTiO
6 DP, which did not show ordering in bulk [
58]. Surprisingly, one can see both long- (XRD) and short-range (TEM–EELS) ordering, driven by a charge disproportionation Ru
4+δ/Ti
4−δ in the SoA film, whereas the (SrRuO
3)
1/(SrTiO
3)
1 SL does not show them. In order to promote the charge transfer and the resulting Ru
5+/Ti
3+ ordering, one most likely has to further tune the processing conditions such as oxygen pressure, deposition time, temperature, which in this case were not optimized for Sr
2RuTiO
6 DP.
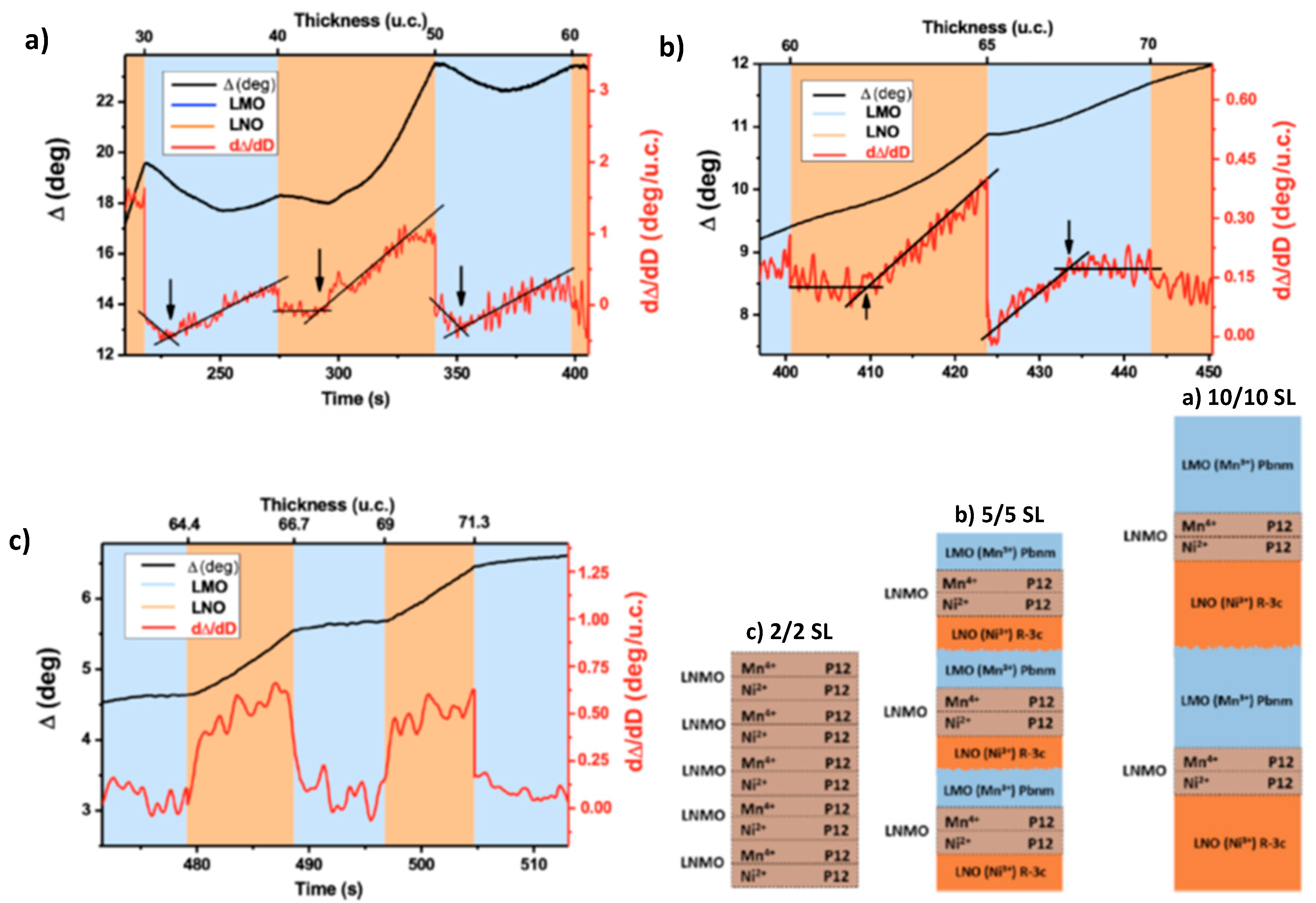
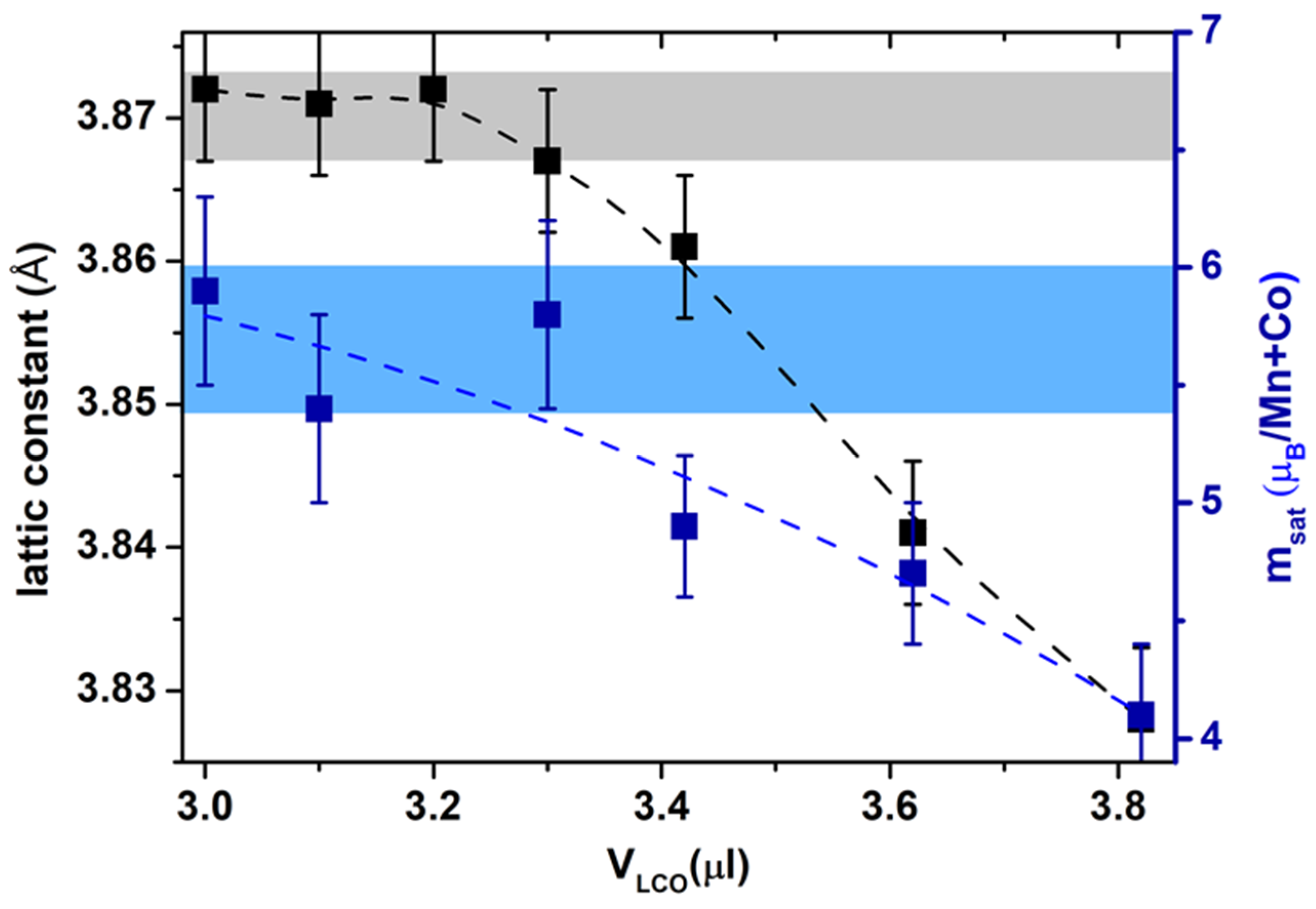
Finally, the presented Layer-by-Layer growth route shows an opportunity to further tune the degree of B-place order by a controlled “layered” stoichiometry, i.e., by the LMO/LCO precursor ratio. Results show that the alternating deposition of the ABO/AB’O atomic layers, with pauses between the LCO and LMO pulses that allow atoms to completely fill the terraces, is promising and likely to suppress the formation of point defects as well as antiphase boundaries. Further research is necessary to optimize the LL growth and stoichiometry control as there were still observed discrepancies between the short-and long-range B-site ordering in the LL-grown LCMO double perovskite films. Particularly, TEM and EELS measurements as well as magnetization (Ms = 6 µB/f.c., Hc = 6 kOe) confirm the high degree of short-range order and give no evidence of APB. However, this does not fit the degree of long-range ordering, quantified by the intensity of the (½ ½ ½) peak in XRD patterns. It is likely that the latter cannot be viewed as a reliable criterion and thus deserves further clarification.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










