1. Introduction
The development of rechargeable magnesium batteries has gained a lot of interest in recent years, mainly due to the advantageous use of magnesium metal anode. Its high volumetric capacity (3833 mAh cm
−3), possible non-dendritic deposition [
1], natural abundance, and geographical distribution make it one of the most promising alternatives to current lithium-ion batteries. The first Mg-battery prototype was proposed by Aurbach et al. [
2], and since then, many improvements have been made on all aspects of the battery system, including anode, cathode, and electrolyte [
3,
4]. The discovery and development of non-nucleophilic electrolytes opened a promising field of magnesium batteries combined with organic or sulfur-based cathodes [
5,
6]. Even though redox-active polymers or sulfur/carbon composites offer attractive energy storage systems, their low volumetric energy density neutralizes one of the most important advantages of the magnesium metal anode, namely, its high volumetric energy density. The use of inorganic materials with high gravimetric density would be beneficial to improve overall volumetric energy density, but reversible magnesium insertion into inorganic phases is limited to only a few known examples, mainly to sulfide-based materials [
2,
7,
8]. Major challenges with magnesium insertion into inorganic materials are several: (i) desolvation of Mg
2+ cation, (ii) structural stability, and (iii) low magnesium ion mobility within the structure in many of the proposed materials [
9]. Chevrel phases, presented in the initial research, offer excellent reversibility and unusually high ion mobility [
10] but their low capacity and low voltage limit their potential use.
The work on layered TiS
2 and spinel-type Ti
2S
4 reported by Nazar’s group, employed the use of sulfides to lessen the interactions between the host and Mg
2+ ion [
7,
8]. At 60 °C and C/20 rate, they achieved approximately 160 mAh g
−1 for both materials. However, at 20 °C, Tao et al. fabricated TiS
2 nanotubes and achieved higher initial capacities but observed severe capacity fade. Use of selenides such as TiSe
2 and WSe
2 improved stability at the cost of lower theoretical specific capacity [
11,
12]. Although chalcogenide-based materials show promising electrochemical behavior, their theoretical energy density remains relatively low for wide commercial acceptance. Thus, the development of high-capacity and high-voltage magnesium cathodes is still a major obstacle toward practical magnesium batteries with high volumetric energy density.
Several different transition metal oxides have been proposed as potential high voltage cathode materials for magnesium batteries [
13]. Among them, manganese oxides are particularly interesting due to their high potential and theoretical capacity (616.6 mAh g
−1 based on MnO
2 molecular weight and considering two-electron reaction), structural variety, and ability to accommodate highly polarizable Mg cations. Theoretical work published by Ling et al. [
14] suggests a possible magnesium insertion into the various manganese oxide polymorphs. Ling and coworkers predicted that the spinel phase and (Mg)Mn
2O
4 isostructural to (Ca)Fe
2O
4 are potential candidates for reversible magnesium intercalation.
Apart from the theoretical work, several groups have used different manganese oxides as insertion host structures for magnesium cations. Sinha et al. [
15] showed electrochemical magnesium insertion in LiMn
2O
4 in aqueous media using cyclic voltammetry measurements. In this work, the anodic and cathodic peaks corresponding to the removal of Li
+ ions during the first cycle and subsequent insertion/disinsertion of Mg
2+ ions are clearly visible. Yuan et al. [
16] investigated intercalation of polyvalent anions in λ-MnO
2, prepared by simple acid treatment of LiMn
2O
4. Magnesium insertion was performed from 0.5 M MgCl
2 aqueous solutions and resulted in a high capacity (545 mAh g
−1) at a current density of 13.6 mA g
− [
1]. The formation of MgMn
2O
4 was proposed from characterization of the sample with inserted magnesium using XPS and powder X-ray diffraction spectroscopy.
A more detailed mechanism study of magnesium intercalation into acid-leached spinel MnO
2 host was performed by Kim and coworkers [
17]. X-ray diffraction characterization pointed out the formation of λ-MnO
2 during charge and formation of tetragonally distorted spinel lattice during discharge. The structure was visualized with ABF STEM, and the Mn reduction was confirmed with XAS measurements. Additional evidence of magnesium insertion into the structure was obtained by
25Mg NMR, where Mg
2+ was found to be in close proximity to paramagnetic centers (Mn
4+ and Mn
3+) in the structure. The formation of Mg
2+ indicates structural changes that were observed [
11,
18] as the formation of a rock salt (Mn
3O
4) phase at the surface.
Despite several studies, a detailed electrochemical and structural comparison of possible magnesium insertion from electrolytes with a different coordination of magnesium is missing. In this work, we discuss magnesium insertion into delithiated manganese spinel in three different electrolytes that possess different solvation properties for magnesium. Magnesium insertion and its reversibility have been studied using different electrochemical techniques. The differences in the capacity fading were thoroughly evaluated with TEM using EDX and EELS. The differences between electrolytes and the associated crystallographic changes are discussed.
3. Results
Commercial LiMn
2O
4 was taken as a starting material without any pretreatment in order to keep the initial cubic spinel structure consistent. Electrochemical delithiation using cycling voltammetry (
Figure 1) in 0.1 M water solution of Mg(NO
3)
2 shows typical Li anodic double peaks in the first cycle [
15] at a potential ~0.7 V and ~0.9 V vs. Ag/AgCl reference electrode (~3.3 V and ~3.5 V vs. magnesium metal). In the subsequent reduction process, a low signature of Li insertion is clearly seen. In the second anodic polarization, this Li feature is absent. The estimated concentration of Li
+ cations in electrolyte is in the range 5.5 × 10
−6 M, which is several orders of magnitude lower compared to pristine Mg electrolyte solution. Upon further cycling, only peaks at 0.25 V and −0.01 V vs. Ag/AgCl reference electrode are present, which indicates potential rearrangements in the structure (
Figure 1a). During the first cycles, anodic and cathodic peaks in the cycling voltammogram (
Figure 1a) are increasing and stabilize around 20th cycle.
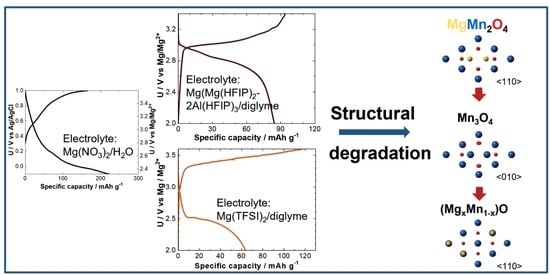
The galvanostatic cycling in 1 M Mg(NO
3)
2 in a water solution (
Figure 1b) shows high electrochemical activity of the initial LiMn
2O
4. The first oxidation proceeds at a higher voltage compared to the following oxidation processes. This can be correlated to the electrochemical removal of Li from the LiMn
2O
4 structure. While capacity during the oxidation process in the first cycle corresponds to almost complete Li removal (obtained capacity is close to theoretical), the following reduction process exhibits an excess of capacity. Higher capacity during discharge in the first cycle is primarily due to the formation of a double-layer capacitance at the high-surface-area carbon black used in this study and partially due to degradation processes. The electrochemical activity observed during the oxidation process is almost fully reversible; however, the system exhibits large polarization and a relatively fast capacity fading.
Insertion of Mg from water-based electrolyte clearly shows that Mg can be inserted into the spinel structure from electrolytes where the Mg solvation is weak [
21,
22]. Since Mg metal cannot be used in water-based electrolytes, the use of non-aqueous electrolytes was evaluated. Mg insertion into manganese spinel from electrolytes containing chlorides (mixture of Mg(TFSI)
2 and MgCl
2 in TEGDME:DOL) was not possible. Galvanostatic cycling in electrolyte without chloride that contained only Mg(TFSI)
2 dissolved in diglyme resulted in much lower initial specific capacity compared to an aqueous counterpart (
Figure 2). Capacity in the first cycle was 65 mAh g
−1, a slightly lower value, as it was obtained in the dual electrolyte system [
23]. Here, the well-defined plateau of Mg insertion at approximately 2.5 V vs. Mg/Mg
2+ counter electrode in the first cycle is in agreement with the cyclic voltammetry measurements in water-based solution. However, in the second cycle, only a small part of Mg can be removed at a potential value between 3.2 and 3.3 V vs. Mg counter electrode. Following insertion proceeds at an approximately 150 mV lower potential compared to that of the first cycle. Increasing polarization indicates a problem with the passivation of the magnesium metal, which was used as a negative electrode. Capacity fading during the initial cycles is very fast. It can be correlated either with the increase in the polarization during initial cycles or with the structural rearrangements in the spinel structure.
Demonstrated galvanostatic cycling in Mg(TFSI)
2/diglyme electrolyte suggests possible partially reversible Mg insertion from the non-aqueous electrolyte. Nevertheless, the insertion is not possible in chloride-containing electrolyte due to the formation of MgCl
+. Recent reports on use of bulky weakly coordinating salts, such as fluorinated alkoxyaluminates, indicated their benefits for the Mg-ion battery system, providing high conductivity and high efficiency of Mg plating [
24]. The chosen electrolyte was shown to be also suitable in a combination with the manganese oxides [
19]. Due to the nature of the anion, the Mg
2+ desolvation is easier, making it less likely for solvated ion to try to be inserted into the structure. Galvanostatic cycling in 0.25 M Mg(HFIP)
2-2Al(HFIP)
3/diglyme electrolyte (
Figure 3a,b) showed excellent cycling performance with a very low polarization in the formation cycles. The observed voltages are near theoretical values for the Mg insertion. Over time, the polarization gradually increases, but the increase is significantly slower than that observed in Mg(TFSI)
2/diglyme electrolyte.
In part, it can be explained with the structure of the HFIP electrolyte, which utilizes bulky weakly coordinating anion [
25]. As a result, the desolvation of Mg
2+ ions is easier, making it less likely for solvated ion to intercalate into the structure. One may speculate that the softer character of anion lessens its interactions with the surface of the particle and may influence the rate of degradation, as was confirmed with Li-ion batteries [
26,
27,
28]. Unfortunately, there is no available research, yet, to elucidate these phenomena in Mg batteries, except a few brief notes, as in the paper on birnessite by Sun et al. With an XPS analysis, they found strongly bonded TFSI
- anions on the surface of the cathode, concluding that the limiting factor for Mg intercalation is also the disruption of the ion pairing of the electrolyte salt in addition to the formation of thermodynamically favorable oxides [
29].
One of the concerns that come with the use of non-aqueous electrolytes in the laboratory cells is related to the Li concentration in an electrolyte after its removal from the LiMn
2O
4 structure. Here, the amount of used electrolyte is much lower compared to that in a beaker cell. Due to a lower amount of the electrolyte, Li concentrations can reach up to 0.28 M, assuming a complete delithiation of the initial LiMn
2O
4 material. Hence, a better electrochemistry can be attributed to the supporting role of present Li ions. To rule this assumption out, ICP-OES measurements on the fully magnesiated samples were performed. In both non-aqueous electrolytes, the Li content was smaller than Mg, although the amounts were not negligible (
Table 1).
ICP results are consistent with electrochemistry in both non-aqueous electrolytes. Electrochemical extraction in Mg(TFSI)2/diglyme electrolyte showed relatively good electrochemical extraction of Li with limited insertion of Mg. The results obtained for 0.25 M Mg(HFIP)2-2Al(HFIP)3/diglyme electrolyte corroborate with the electrochemistry, since approximately 56 at.% of Li sites were substituted by Mg. This corresponds to approximately 83 mAh.g−1 capacity, which was observed in the formation cycles.
The Mg insertion and distribution in manganese spinel particles was assessed by STEM-EDX (
Figure 4). Analysis of the sample cycled in the Mg(NO
3)
2 electrolyte (
Figure 4a) confirmed that the anodic peak observed in the cyclic voltammogram (
Figure 1a) corresponds to the magnesiation process. Although the Mg STEM-EDX maps (
Figure S1) from all three samples seem to have a uniform contrast throughout the investigated particles, a closer look at Mn/Mg count ratios taken at the edge and in the interior areas of the particles reveals a different situation (
Figure 4a–c, areas marked by the red lines). In all samples, the outer edges of the particles demonstrate a lower Mn/Mg count ratio (
Figure 4d), which corresponds to a higher amount of Mg. This indicates a presence of core-shell structure with the magnesiated spinel oxide in the shell, and most likely, the remaining LiMn
2O
4 in the core is present. Both samples cycled in non-aqueous electrolytes show a significant difference between core and shell Mn/Mg ratios (
Figure 4d) with the Mg(HFIP)
2 − 2Al(HFIP)
3/diglyme-cycled sample demonstrating the highest Mg amount in the shell area. In case of aqueous electrolyte-cycled sample, STEM-EDX map taken from the sample cycled to 100 cycles showed rather erroneous homogenous Mg distribution due to a complete microstructure deformation described later in the text. Therefore, the EDX map shown in
Figure 4a was taken from the sample after five cycles in order to obtain a more comparable Mg distribution picture in pristine microstructure.
Cathodes materials’ microstructure and crystal structure changes on the atomic level upon cycling in the three different electrolytes are shown in
Figure 5. Here, the Mg(NO
3)
2/diglyme-cycled sample microstructure has the most damaged appearance (
Figure 5a). The spinel material has degraded upon 100 cycles from larger up to 300 nm single crystal particles into agglomerates of nanoparticles with disturbed crystallinity and sizes of about 5–10 nm (
Figure 5a,d and
Figure S2). The Mg(TFSI)
2/diglyme-cycled sample that showed a very rapid capacity reduction in the first five cycles and failed after only 15 cycles retained a single crystal formation. The microstructure at the edges of the particle appears significantly etched and porous (
Figure 5b). The Mg(HFIP)
2 − 2Al(HFIP)
3/diglyme-cycled material, where much better electrochemical behavior with more linear capacity drop throughout the cycling was observed (
Figure 3b), revealed the least deformed microstructure with not many visible pores or defects after 60 cycles (
Figure 5c).
Thorough inspection of samples at the atomic resolution level showed similar patterns of structural degradation of LiMn
2O
4-type spinel structure for all applied electrolytes (
Figure 5d–f). Atomic columns intensity patterns, such as in the areas marked by red and green dotted lines in
Figure 5e,f, were observed in the large amounts in all three samples throughout the electron transparent areas. The green dotted lines mark the areas that correspond to the rock salt Mg/Mn mixed structure, while the red ones mark the STEM-HAADF contrast pattern of Mn
3O
4-type spinel.
A more detailed presentation of all three crystal structures, i.e., Mg
xMn
2O
4, Mn
3O
4, and (Mg
xMn
1-x)O, is given in
Figure 6. The STEM-HAADF representative atomic resolution images (
Figure 6a–c) were cut out from the adjacent areas near the edge of the particle in a single micrograph, taken from the sample cycled in aqueous electrolyte for five cycles. As it can be seen in the atomic structure sketches in
Figure 6e and in the intensity profiles of all three structures plotted in
Figure 6d, the changes in the atomic arrangement and interatomic distances in crystal structures can be monitored by observing the so-called Mn diamond configuration [
30], marked by dashed lines in
Figure 6. This configuration is visible in the <010> tetragonal spinel and <110> cubic spinel and cubic rock salt structural projections. In the nominal MgMn
2O
4 structure, the corners of the Mn diamond are occupied by the Mn atoms at 16d positions, while Mg occupies 8a sites. Changes in STEM-HAADF contrast that can be seen within the central part of the Mn diamond (
Figure 6a–c) correspond to the initial Mg spinel structure being transformed into Mn
3O
4 and rock salt structures. In Mn
3O
4, the Mn atoms occupy the 8a positions in diamond configuration that are left open upon Mg leave (
Figure 6b,e2). This is observed as the increased intensity at 8a sites in the center of the diamond configuration compared to that of MgMn
2O
4 (see intensity profiles in
Figure 6d with respective colors). In rock salt, the diamond configuration exhibits only one strong contrast in the central octahedral site (
Figure 6c,e3). Here, the transformation from the normal spinel proceeds via the insertion of the Mg cations into vacant 16c sites followed by the Frenkel defect-mediated shift of Mg from tetrahedral to octahedral sites [
31].
The majority of reported synthesized MgMn
2O
4 spinels possess tetragonal
I41/amd symmetry, where Mn
3+ ions at the octahedral sites are distorted by a cooperative Jahn–Teller effect. Only Truong and coworkers have reported on synthesis and electrochemical cycling of cubic MgMn
2O
4 in Mg(ClO
4)
2/acetonitrile and Mg(NO
3)
2 electrolytes [
18,
30]. In this study, the cubic
Fd-3m LiMn
2O
4 spinel was used as a precursor in order to see the effect of magnesiation on the normal cubic spinel structure. XRD measurements of water-based electrolyte-cycled sample proved to be difficult due to the need for collecting the large amount of cycled material. Due to the specifics of material cycling that was employed in order to receive larger amount of cycled sample, the amount of magnesiated material in XRD measurements was not in accordance with ICP measurements. The latter was performed on a material cycled in smaller amounts in a way described in the experimental section. The observed magnesiated fraction possessed a cubic structure with a smaller unit cell parameter (from
a = 8227 Å in pristine LiMn
2O
4, to
a = 8174 Å in delithiated state, to
a = 8163 Å upon magnesiation) compared to that of the LiMn
2O
4 spinel (
Figure S3), which is in the agreement with recent studies. Therefore, for monitoring the local crystal structure changes after cycling to failure in each of the three electrolytes, the ratio of short (S) to long (L) diagonals of the Mn diamond configuration displayed in
Figure 6 was calculated from the experimental STEM-HAADF images (
Table 2) [
30]. For comparison, the cubic and tetragonal structure end members’ ratios are also given. For the experimental measurements presented in
Table 2, only areas with the MgMn
2O
4-like diamond configuration were considered in the STEM-HAADF images.
The obtained S/L ratio of 0.718 for Mg(NO
3)
2-cycled sample was the closest to the cubic structure values, which is in accordance with XRD measurements. Mg(TFSI)
2/diglyme-cycled sample exhibited the most tetragonal distortion of the magnesiated spinel structure, while the Mg(HFIP)
2 − 2Al(HFIP)
2/diglyme-cycled sample values were around the middle value of two end members, with a slight tendency towards tetragonal. In the case of the latter sample, more Mg was inserted into the delithiated LiMn
2O
4 according to the ICP measurements (see
Table 1), while the former sample showed good delithiation but poorer magnesiation behavior. In all samples, the diamond configuration measured in the areas of the Mn
3O
4 structure had tetragonal S/L ratios (S/L = 0.632), while the S/L ratios from the rock salt structure areas deviated from the expected cubic by approximately 50% towards tetragonal values (S/L = 0.695).
We applied STEM-EELS analysis to explore changes in Mn valence from the surface into the interior of the magnesiated manganese spinel particles upon cycling in three different electrolytes. Due to the variations in the chemical bonding and the environment of a specific atom, so-called chemical shift of the Mn L
2,3 energy-loss edge onset is induced along with modifications in the near-edge fine structure (ELNES) of the EEL spectrum. While the valence numbers can be with some precautions calculated from the Mn L
3/L
2 peaks ratio [
32], chemical shift of the edge onset allows for a quick way to monitor changes in the Mn valence. In order to keep the electron beam damage minimal, fast acquisition of EEL spectra during the line scan was applied.
EEL spectra of Mn L
2,3 edge revealed that in all three samples, the Mn L
2,3 edge onset position experienced a chemical shift, which was observed up to the 15 nm depth into the particle’s interior (
Figure 7). Please note that the positions of the Mn L
3 peaks instead of edge onsets are marked for easier visual differentiation. Relying on peak positions for the determination of true chemical shift would lead to erroneous values due to the differences in ELNES fine structure of the edge, which depends on the Mn atom surrounding in the structure. Both non-aqueous electrolytes-cycled samples (
Figure 7b,c,e,f) show gradual chemical shift towards higher valence states of Mn. The Mg(TFSI)
2-cycled sample has reaches the chemical shift of 0.75 eV within 12.5 nm into particle’s depth on average with edge onset starting at 639.25 eV and finishing at 640 eV, while the Mg(HFIP)
2 − 2Al(HFIP)
3-cycled sample has reached the same chemical shift within 8.72 nm on average. The L
3/L
2 ratios for the area within first 15 nm particle depths in both samples were 2.44 and 2.7, respectively. These values correspond to mixed Mn
2+/Mn
3+ valences, with the Mg(TFSI)
2-cycled sample having more Mn
2+ and the Mg(HFIP)
2 − 2Al(HFIP)
3-cycled sample having more Mn
3+ species [
32]. This is in accordance with the observation of large amounts of Mn
3O
4 and Mg/Mn mixed rock salt structures by STEM-HAADF. The sample cycled in aqueous electrolyte, however, revealed rather random fluctuations of chemical shift of 0.5 eV within the whole analysis area due to the disintegrated microstructure described earlier (
Figure 7a,d). The Mn L
3/L
2 edge onset position was lying between 639.75 eV and 640 eV, while the average L
3/L
2 ratio for the area of first 15 nm particle depths was 3.0. This corresponds as well to the mixture of Mn
2+ and Mn
3+ species; however, it implicates the highest Mn
2+ amount among all three samples. The result corroborates the STEM-HAADF imaging data, where the aqueous sample demonstrated the most disintegrated microstructure.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
