
Structures of a Phosphoryl Derivative of 4-Allyl-2,4-dihydro-3H-1,2,4-triazole-3-thione: An Illustrative Example of Conformational Polymorphism

 , ,
, ,
Abstract
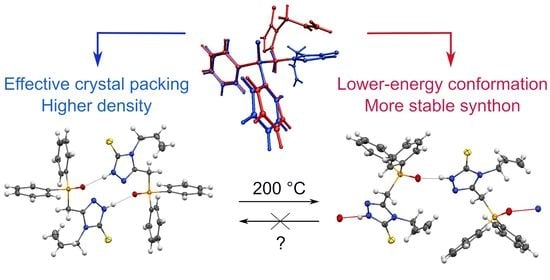
:
1. Introduction
2. Materials and Methods
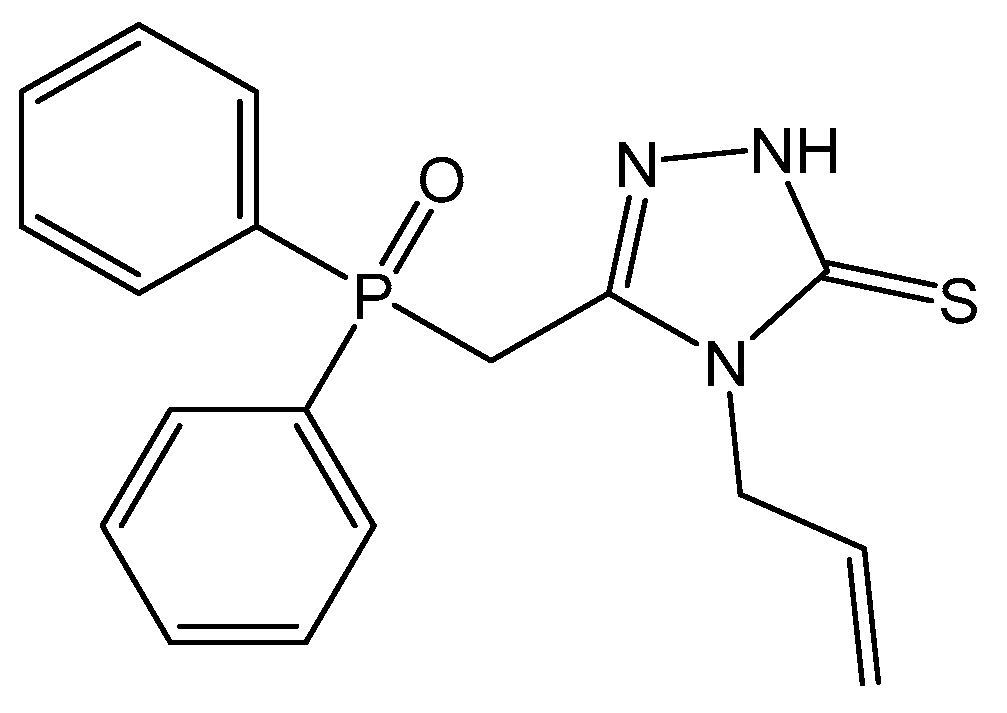
2.1. Synthesis and Crystallization
2.2. Single-Crystal X-ray Crystallography
2.3. Powder X-ray Diffraction Experiments
2.4. Differential Scanning Calorimetry Experiments
2.5. Computational Details
3. Results and Discussion
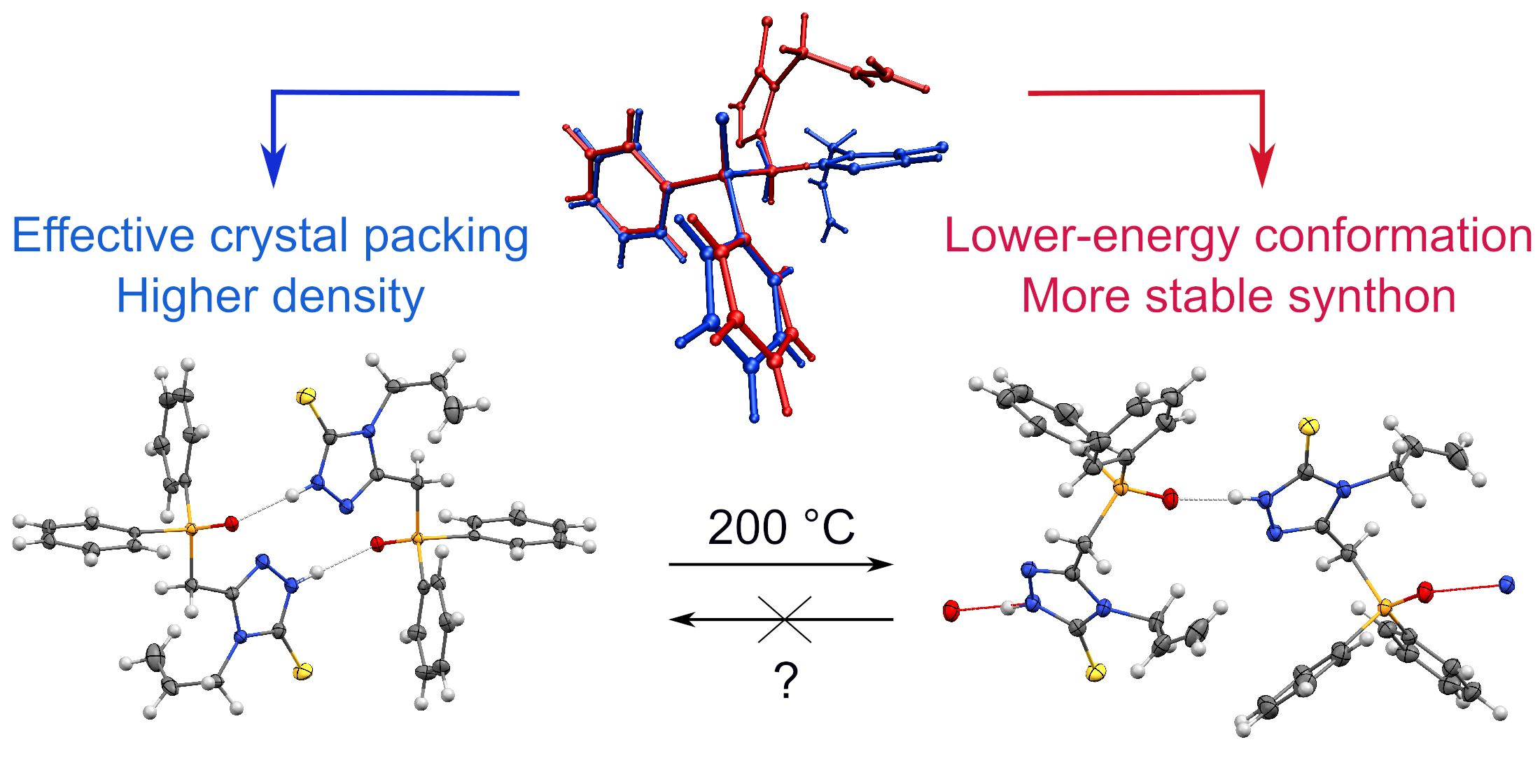
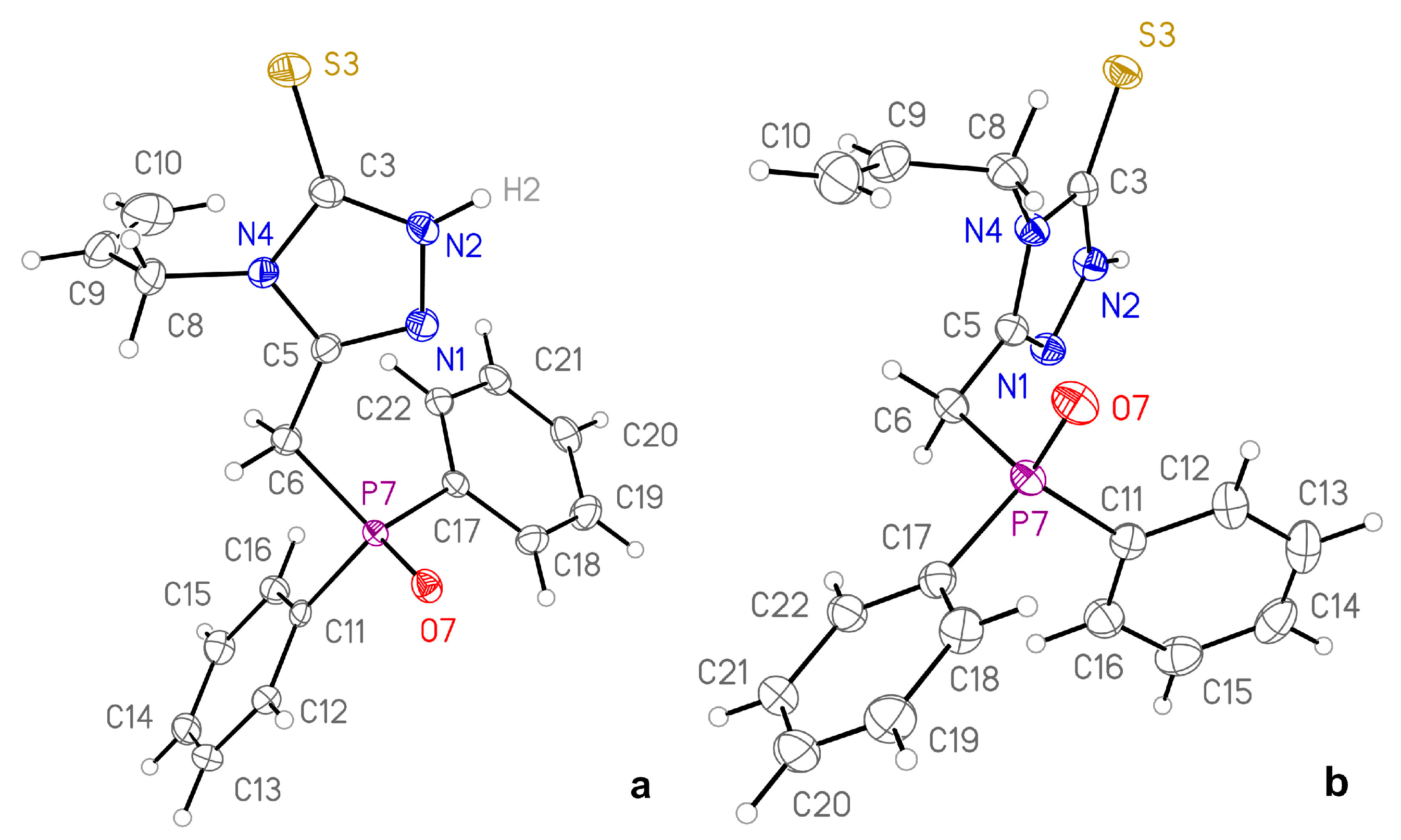
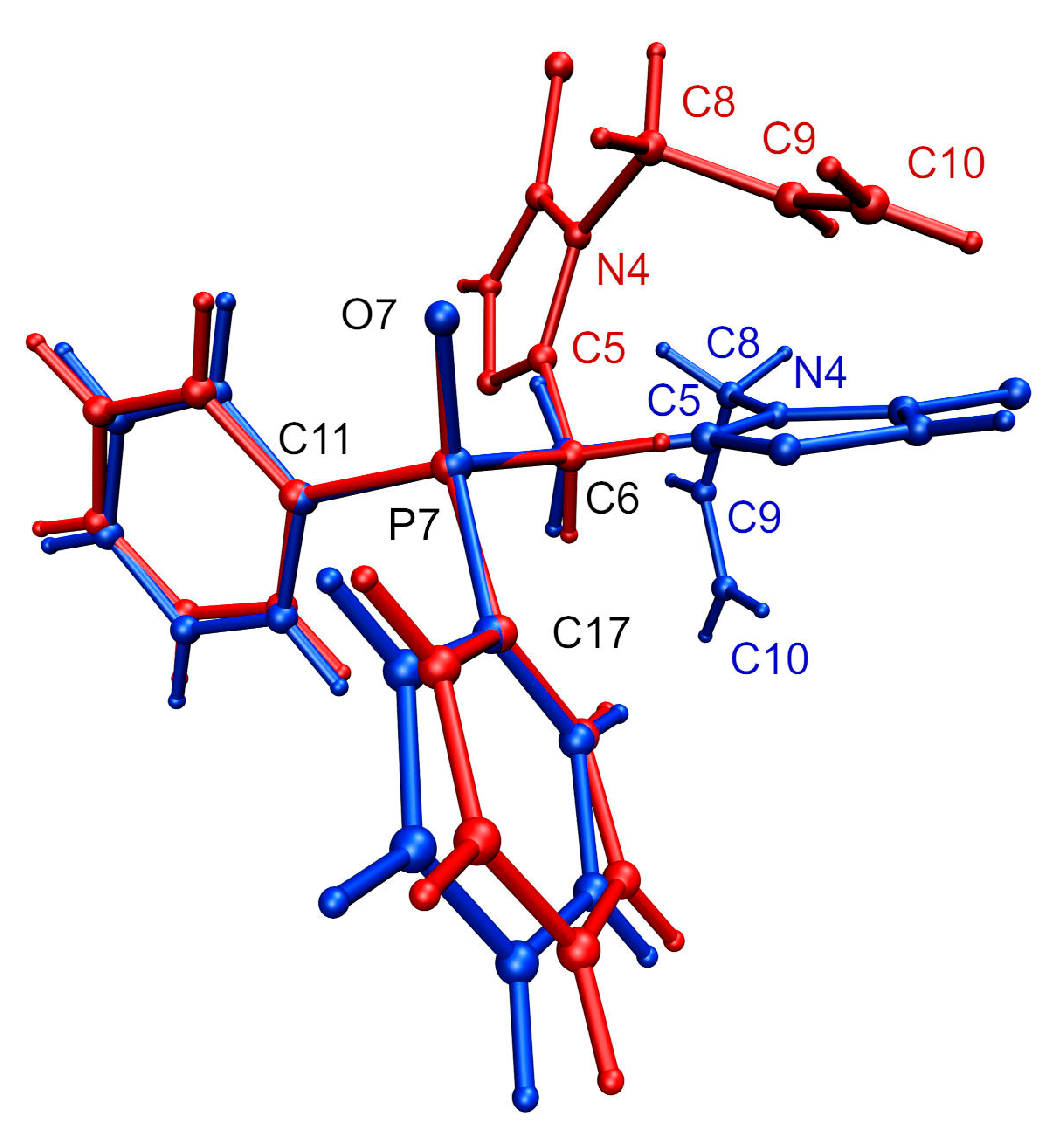
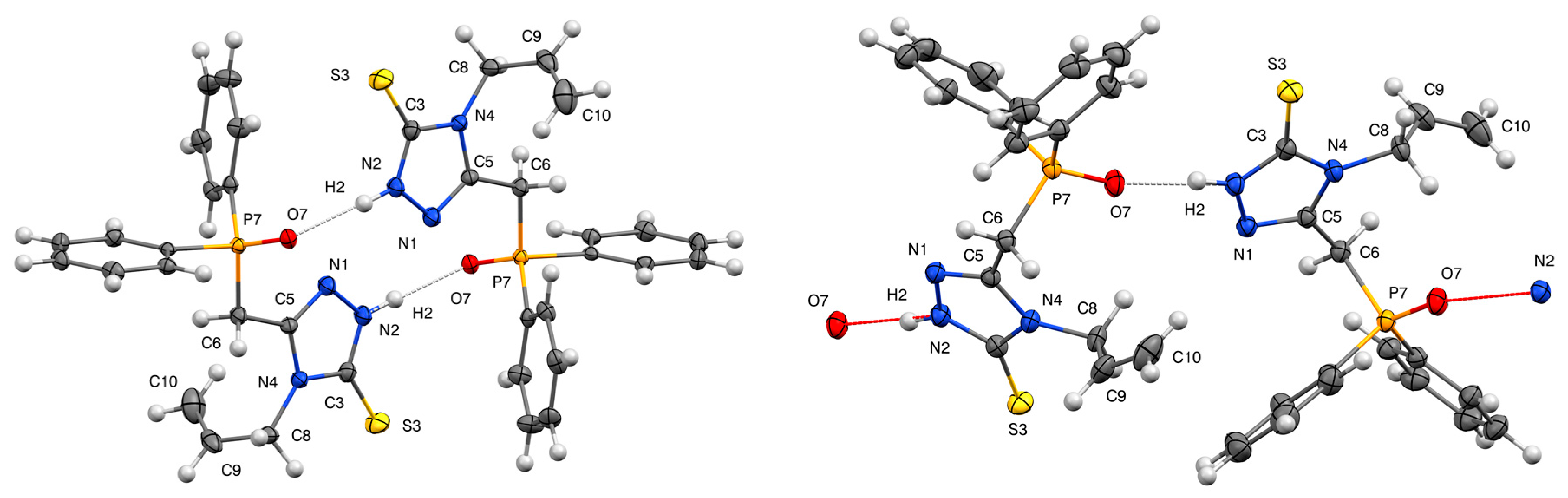
3.1. Molecular Geometry and Crystal Packing
3.2. Ab Initio Calculations
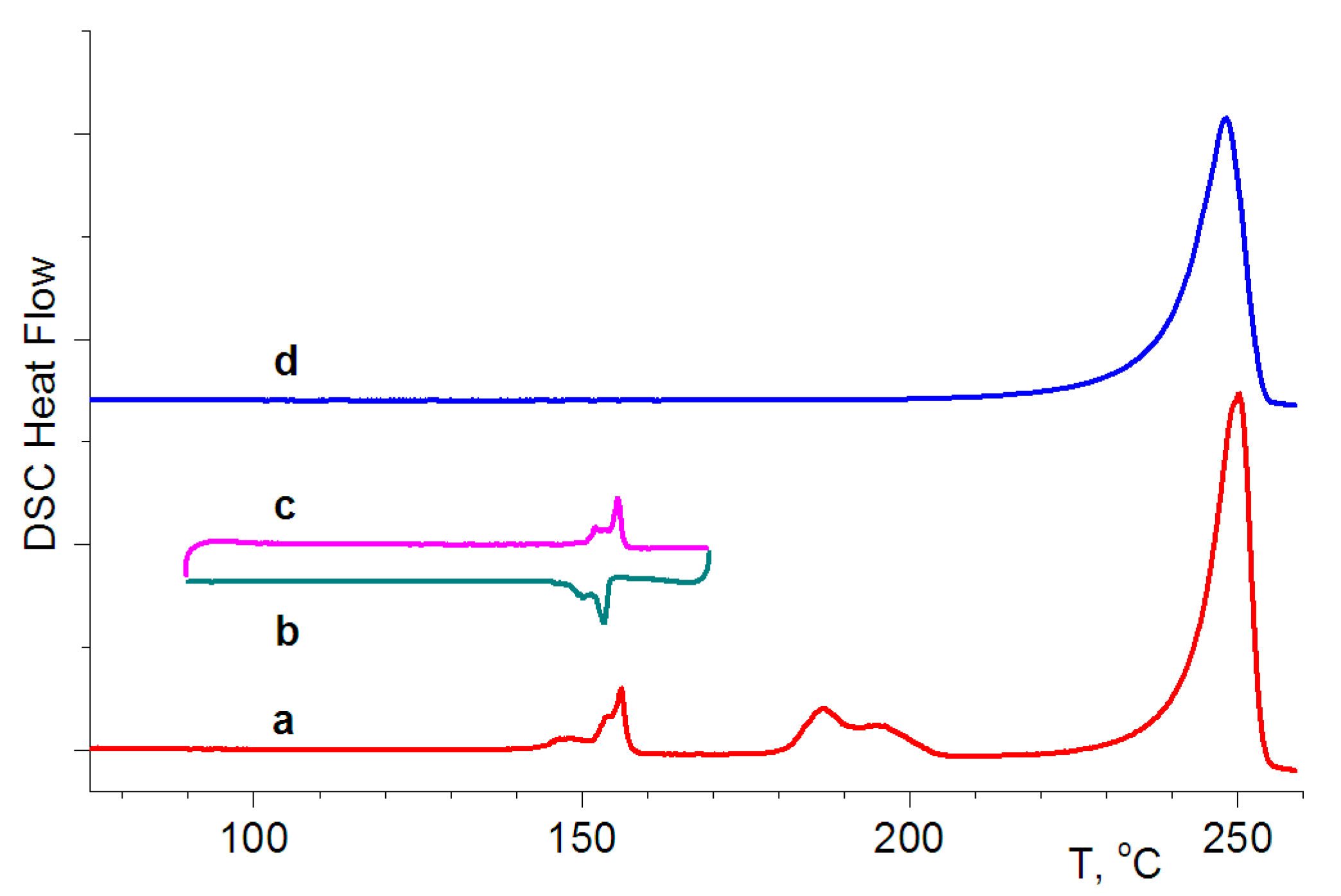
3.3. Differential Scanning Calorimetry Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bernstein, J. International Union of Crystallography monographs on crystallography. In Polymorphism in Molecular Crystals; Oxford University Press: New York, NY, USA; OxfordClarendon Press: New York, NY, USA, 2002; ISBN 0-19-850605-8. [Google Scholar]
- McCrone, W.C. Polymorphism. In Physics and Chemistry of the Organic Solid State; Fox, D., Labes, M.M., Weissberger, A., Eds.; Interscience Publishers: London, UK, 1965; Volume 2, pp. 725–767. [Google Scholar]
- Lee, E.H. A Practical Guide to Pharmaceutical Polymorph Screening & Selection. Asian J. Pharm. Sci. 2014, 9, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Hilfiker, R. (Ed.) Polymorphism in the Pharmaceutical Industry; Wiley-VCH: Weinheim, Germany, 2006; ISBN 978-3-527-31146-0. [Google Scholar]
- Bučar, D.-K.; Lancaster, R.W.; Bernstein, J. Disappearing Polymorphs Revisited. Angew. Chem. Int. Ed. 2015, 54, 6972–6993. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational Polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Jetti, R.K.R.; Boese, R.; Sarma, J.A.R.P.; Reddy, L.S.; Vishweshwar, P.; Desiraju, G.R. Searching for a Polymorph: Second Crystal Form of 6-Amino-2-Phenylsulfonylimino-1,2-Dihydropyridine. Angew. Chem. Int. Ed. 2003, 42, 1963–1967. [Google Scholar] [CrossRef]
- Gavrilova, E.L.; Krutov, I.A.; Valieva, A.A.; Khayarov, K.R.; Samigullina, A.I.; Gubaidullin, A.T.; Shatalova, N.I.; Burangulova, R.N.; Sinyashin, O.G. Synthesis of New Phosphorylated 1,2,4-Triazole-3-Thiones. N,S-Functionalization Methods. Russ. J. Gen. Chem. 2018, 88, 2269–2275. [Google Scholar] [CrossRef]
- Li, C.; Liu, J.-C.; Li, Y.-R.; Gou, C.; Zhang, M.-L.; Liu, H.-Y.; Li, X.-Z.; Zheng, C.-J.; Piao, H.-R. Synthesis and Antimicrobial Evaluation of 5-Aryl-1,2,4-Triazole-3-Thione Derivatives Containing a Rhodanine Moiety. Bioorganic. Med. Chem. Lett. 2015, 25, 3052–3056. [Google Scholar] [CrossRef] [PubMed]
- Celik, G.; Khloya, P.; Vullo, D.; Supuran, C.T.; Sharma, P.K. Benzenesulfonamide Bearing 1,2,4-Triazole Scaffolds as Potent Inhibitors of Tumor Associated Carbonic Anhydrase Isoforms HCA IX and HCA XII. Bioorganic. Med. Chem. 2014, 22, 1873–1882. [Google Scholar] [CrossRef]
- Almasirad, A.; Tabatabai, S.A.; Faizi, M.; Kebriaeezadeh, A.; Mehrabi, N.; Dalvandi, A.; Shafiee, A. Synthesis and Anticonvulsant Activity of New 2-Substituted-5- [2-(2-Fluorophenoxy)Phenyl]-1,3,4-Oxadiazoles and 1,2,4-Triazoles. Bioorganic Med. Chem. Lett. 2004, 14, 6057–6059. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Emami, S.; Foroumadi, A. The Importance of Triazole Scaffold in the Development of Anticonvulsant Agents. Eur. J. Med. Chem. 2016, 109, 380–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, S.A.; Dowd, C.S. Phosphoryl Prodrugs: Characteristics to Improve Drug Development. Med. Chem. Res. 2021. [Google Scholar] [CrossRef]
- Krutov, I.A.; Gavrilova, E.L.; Burangulova, R.N.; Kornilov, S.S.; Valieva, A.A.; Samigullina, A.I.; Gubaidullin, A.T.; Sinyashin, O.G.; Semina, I.I.; Nikitin, D.O.; et al. Modification of Diphenylphosphorylacetic Hydrazide with Thiosemicarbazide and Triazole Units. Russ. J. Gen. Chem. 2017, 87, 2794–2800. [Google Scholar] [CrossRef]
- Bruker. APEX2; Bruker AXS Inc.: Madison, WI, USA, 2006. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Agilent Technologies. CrysAlisPro; Agilent Technologies Ltd.: Santa Clara, CA, USA, 2012. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Single-Crystal Structure Validation with the Program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Bruker. DIFFRAC Plus EVA.; Bruker AXS Inc.: Karlsruhe, Germany, 2005. [Google Scholar]
- Coelho, A.A. TOPAS and TOPAS-Academic: An Optimization Program Integrating Computer Algebra and Crystallographic Objects Written in C++. J. Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Vilela Oliveira, D.; Laun, J.; Peintinger, M.F.; Bredow, T. BSSE-correction Scheme for Consistent Gaussian Basis Sets of Double- and Triple-zeta Valence with Polarization Quality for Solid-state Calculations. J. Comput. Chem. 2019, 40, 2364–2376. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Gatti, C.; Saunders, V.R.; Roetti, C. Crystal Field Effects on the Topological Properties of the Electron Density in Molecular Crystals: The Case of Urea. J. Chem. Phys. 1994, 101, 10686. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 19.10.12); TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Ananyev, I.V.; Karnoukhova, V.A.; Dmitrienko, A.O.; Lyssenko, K.A. Toward a Rigorous Definition of a Strength of Any Interaction between Bader’s Atomic Basins. J. Phys. Chem. A 2017, 121, 4517–4522. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A. How Reliable Are Intermolecular Interaction Energies Estimated from Topological Analysis of Experimental Electron Densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Jayatilaka, D.; Grimwood, D.J. Tonto: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography. In Computational Science—ICCS 2003; Sloot, P.M.A., Abramson, D., Bogdanov, A.V., Gorbachev, Y.E., Dongarra, J.J., Zomaya, A.Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; Volume 2660, ISBN 978-3-540-40197-1. [Google Scholar]
- Bondi, A. Van Der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Kessler, M.; Luo, J.; Motherwell, W.D.S.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of Crystallographically-Derived Molecular Geometry Information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Kitaigorodskii, A.I. Organic Chemical Crystallography; Consultant’s Bureau: New York, NY, USA, 1961; ISBN 978-0-592-01213-1. [Google Scholar]
- Gidaspov, A.A.; Zalomlenkov, V.A.; Bakharev, V.V.; Parfenov, V.E.; Yurtaev, E.V.; Struchkova, M.I.; Palysaeva, N.V.; Suponitsky, K.Y.; Lempert, D.B.; Sheremetev, A.B. Novel Trinitroethanol Derivatives: High Energetic 2-(2,2,2-Trinitroethoxy)-1,3,5-Triazines. RSC Adv. 2016, 6, 34921–34934. [Google Scholar] [CrossRef] [Green Version]
- Saifutiarova, A.E.; Karnoukhova, V.A.; Gulakova, E.N.; Fedorova, O.A.; Fedyanin, I.V. Molecular Structures and Crystal Packings of Styryldiazine. J. Struct. Chem. 2021, 62, 527–536. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1a | 1b |
|---|---|---|
| Chemical formula | C18H18N3OPS | |
| M | 355.38 | |
| Single-crystal XRD | ||
| CCDC number | 2104042 | 2104043 |
| Temperature, K | 150 | 100 |
| Crystal shape, color, size (mm) | colorless block 0.08 × 0.47 × 0.80 | colorless prism 0.06 × 0.11 × 0.16 |
| Crystal system, space group | Monoclinic, P 21/c | Monoclinic, P 21/c |
| Z, Z′ | 4, 1 | 4, 1 |
| Unit cell dimension | a = 12.9204(13) Å b = 9.3989(9) Å c = 14.3269(14) Å β = 103.436(5) ° | a = 12.9164(3) Å b = 12.6337(2) Å c = 12.0704(3) Å β = 116.155(3) ° |
| Volume, Ǻ3 | 1692.2 (3) | 1767.99 (8) |
| dcalc, g cm−3 | 1.395 | 1.335 |
| Radiation type | MoKα | CuKα |
| μ (mm−1) | 0.296 | 2.557 |
| 2θ range for data collection (°) | 5.8–53.7 | 7.6–152.2 |
| Measured reflections | 22,997 | 20,923 |
| Independent reflections | 3614 | 3654 |
| R(int) | 0.0479 | 0.0515 |
| No. of parameters | 221 | 221 |
| Reflections with I 2σ(I) | 3026 | 3336 |
| R1/wR2 [I > 2σ(I)] | 0.0359/0.0876 | 0.0385/0.1079 |
| R1/wR2 (all reflections) | 0.0458/0.0932 | 0.0417/0.1108 |
| GooF = S | 1.051 | 1.059 |
| ρmax/ρmin (e Ǻ−3) | 0.379/−0.329 | 0.390/−0.427 |
| Powder XRD (Pawley method) | ||
| Temperature, K | 298 | 298 |
| Unit cell dimension | a = 12.9709(19) Å b = 9.4680(14) Å c = 14.558(2) Å β = 103.7143(18) ° | a = 12.9645 (15) Å b = 12.6921 (16) Å c = 12.1837 (15) Å β = 114.7107 (18) ° |
| Volume, Ǻ3 | 1736.9(5) | 1821.2 (4) |
| dcalc, g/cm3 | 1.359 | 1.296 |
| R(wp) | 0.046 | 0.037 |
| 1a | 1b | |
|---|---|---|
| φ1 (O7-P7-C11-C12) | 21.93 (15) | 11.72 (15) |
| φ2 (O7-P7-C17-C18) | 41.22 (16) | 36.49 (14) |
| φ3 (O7-P7-C6-C5) | 65.16 (15) | −60.78 (12) |
| φ4 (P7-C6-C5-N4) | −170.98 (13) | 84.59 (16) |
| φ5 (C5-N4-C8-C9) | −84.0 (2) | 63.7 (2) |
| φ6 (N4-C8-C9-C10) | −8.8 (3) | −134.75 (18) |
| Experimental | Crystal DFT a | |||
|---|---|---|---|---|
| 1a | 1b | 1a | 1b | |
| N-H b, Å | 1.040 | 1.043 | 1.040 | 1.043 |
| N⋯O, Å | 2.754 (2) | 2.6686 (16) | 2.722 | 2.606 |
| H⋯O, Å | 1.727 | 1.634 | 1.689 | 1.569 |
| N-H⋯O,° | 168.9 | 170.8 | 171.4 | 172.3 |
| ρ(r), e A−3 | 0.307 | 0.393 | ||
| ∇2ρ(r), e A−5 | 3.33 | 4.09 | ||
| EEML, kcal/mol | 15.2 | 21.0 | ||
| 1a | 1b | |
|---|---|---|
| Cohesive energy (Ecog) | ||
| DFT | 49.7 | 46.6 |
| Lattice energy (Elatt) | ||
| DFT | 60.4 | 52.0 |
| CE-B3LYP | 56.2 | 45.8 |
| EML | 49.7 | 46.6 |
| Synthon bonding energy (dimer/chain) | ||
| DFT | 15.9 | 18.0 |
| CE-B3LYP | 12.7 | 14.2 |
| EML | 16.3 | 23.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedyanin, I.V.; Samigullina, A.I.; Krutov, I.A.; Gavrilova, E.L.; Zakharychev, D.V. Structures of a Phosphoryl Derivative of 4-Allyl-2,4-dihydro-3H-1,2,4-triazole-3-thione: An Illustrative Example of Conformational Polymorphism. Crystals 2021, 11, 1126. https://doi.org/10.3390/cryst11091126
Fedyanin IV, Samigullina AI, Krutov IA, Gavrilova EL, Zakharychev DV. Structures of a Phosphoryl Derivative of 4-Allyl-2,4-dihydro-3H-1,2,4-triazole-3-thione: An Illustrative Example of Conformational Polymorphism. Crystals. 2021; 11(9):1126. https://doi.org/10.3390/cryst11091126
Chicago/Turabian StyleFedyanin, Ivan V., Aida I. Samigullina, Ivan A. Krutov, Elena L. Gavrilova, and Dmitry V. Zakharychev. 2021. "Structures of a Phosphoryl Derivative of 4-Allyl-2,4-dihydro-3H-1,2,4-triazole-3-thione: An Illustrative Example of Conformational Polymorphism" Crystals 11, no. 9: 1126. https://doi.org/10.3390/cryst11091126
APA StyleFedyanin, I. V., Samigullina, A. I., Krutov, I. A., Gavrilova, E. L., & Zakharychev, D. V. (2021). Structures of a Phosphoryl Derivative of 4-Allyl-2,4-dihydro-3H-1,2,4-triazole-3-thione: An Illustrative Example of Conformational Polymorphism. Crystals, 11(9), 1126. https://doi.org/10.3390/cryst11091126