1. Introduction
Lanthanum transition metal perovskites (of chemical formula LaBO
3) have recently attracted tremendous attention due to their pivotal importance from both fundamental and practical points of view. The B-site transition metal coordinates with the six nearest O atoms, forming an octahedral environment in the bulk of the perovskite structure. On the surface layer, the BO
6 octahedron lacks an apical O atom and exposes the B site. This active site plays an essential role in facilitating a variety of catalytic reactions in heterogeneous catalysis [
1] and electro-catalysis [
2]. Therefore, it is beneficial to develop fabrication methods for oxide perovskites with increased specific surface areas and abundant active sites [
3]. Cobalt ions can adopt some different oxidation states. On that account, lanthanum cobaltite perovskite LaCoO
3 (LCO) endows flexibility in regulating the valence distribution of the Co ion by hole doping or electron doping [
4]. This perspective has prompted a burgeoning interest in and extensive studies on the tunable electronic structure of LCO. The objective is to scale down the perovskite structures into the nanoscale regime and incorporate rare earth ions with various valences into these structures to enhance their catalytic activity and expand their applications [
5].
The physics of pristine LCO and metal-doped counterparts show an abundance of peculiar properties. The interpretation for the crystal structure of LCO has been a rhombohedral
R-3c symmetry [
6,
7], where a Jahn-Teller distortion [
8] is absent. A monoclinic symmetry of space group
I2/a [
7,
9] is also possible, of which the spin state of the Co
3+ ion is Jahn-Teller active. Accordingly, the rhombohedral lattice preserves one Co–O distance, but the monoclinic structure exhibits a differentiation of Co–O bond lengths [
9]. There has long been a discussion on the spin states of these compounds, which also involves a conventional picture with the splitting of t
2g and e
g orbitals and the double-exchange model with the e
g band as the conduction channel [
8,
10,
11,
12,
13,
14,
15,
16,
17]. Rare earth metal substitution that alters the oxidation state of Co can also modulate the occupancy of the
eg-like orbitals and thus the nature of the metal-oxygen bond. Earlier research works on hole-doped LCO found a quasi-stable spin state crossing the Fermi level [
15] and that the e
g states interact with O 2
p states, spreading over a wide energy range [
18]. These findings elicit the same line of inquiry on cerium doping to understand the changes in the metal-oxygen bonding properties associated with the electron-doped LCO.
Previous studies on the Ce-doped LCO system have mainly explored the magnetic properties but have paid little attention to the metal-oxygen bonding [
19,
20,
21,
22]. These works demonstrate that Ce-doping can induce and stabilize the intermediate spin or low-spin high-spin configurations and hint that a high content of dopants may increase the occupancy of the e
g-like states. However, up-to-date studies remain scarce on the possible phase transformation of Ce-doped LCO with high Ce contents and the connection with changes in the local structure. In particular, the details of how Ce-doping modifies the atomic interactions of metal-oxygen bonding, especially the La–O bonding, are still largely unexplored.
In this work, we report on the structure and local structure of nanoscale La1−xCexCoO3, where x is the nominal degree of substitution and x = 0, 0.2, and 0.4. We use the Rietveld refinement method to fit various models of the main lattice and valence and fraction of Ce dopants to the experimental X-ray diffraction (XRD) data. For clarity, such notations as La1−sCesCoO3 or will refer to the main lattice, depending on the form of Ce dopants. The data do not precisely point out to what extent Ce can be present in the LCO lattice. Nevertheless, we find Ce insertion into the main phase plausible and the Ce4+/Co2+ ions more favorable than Ce3+ ions as dopants. Our estimates of the Ce valence of total Ce species in the sample are achievable based on X-ray absorption near edge structure (XANES) data at Ce L3-edge. We illuminate the noticeable change of XANES spectra at La L3-edge as an effect of Ce-doping by conducting the topological analysis of the experimental electron densities and mapping the corresponding electronic energy distributions. We also present an in-depth investigation on the extended X-ray absorption structure (EXAFS) by fitting to data using the models built from the Rietveld refinement results. Building fitting models to EXAFS spectra of these samples is highly challenging due to three issues: (1) each sample involves two scattering phases, (2) the secondary scattering phase, realized as a Co3O4-like phase, has two inequivalent adsorbing sites, and (3) handling dopants, which is an arcane manipulation. Hence, we needed a sophisticated strategy in EXAFS data analysis, as discussed in the following sections.
3. Data Analysis
Quantitative analysis Rietveld refinement of the XRD data was carried out using the Marquardt least-squares algorithm, implemented in the Materials Analysis Using Diffraction (MAUD) software package (Luca Lutterotti, Trento, Italy) [
25]. We performed Rietveld full-spectrum fitting for various models to determine backgrounds, unit-cell parameters, weight percentages, and microstructure for the individual phases present in each sample. Regarding the main lattice, we considered six initial models, including three different perovskite structures and two forms of dopants. The three structures are rhombohedral, monoclinic, and cubic, described by the space groups
R-3c: R (No. 167) [
26],
I12/a1 (No. 15) [
7], and
Pm-3m (No. 221) [
27], respectively. The two cases of dopants were Ce
3+ and Ce
4+. For the case Ce
4+, the cobalt ions were set to adopt Co
2+ for charge compensation. In addition, we introduced
s, the fraction of cerium dopants, into the main lattice as La
1−sCe
sCoO
3 or
and performed a series of fits, altering the value of
s from 0 to 1 by an increment of 0.05. We also considered a secondary phase as cubic Co
3O
4 (space group
Fd-3m, No. 227) [
28] for the
x = 0 sample, and as cubic CeO
2 (space group
Fm3m, No. 225) [
29] for the
x = 0.2 and
x = 0.4 samples. The quality of Rietveld fits was evaluated based on both the visual inspection of observed and calculated patterns and the comparison of the values of discrepancy indices. The most straightforward index is the weight profile
R-factor (
Rwp) [
30].
X-ray absorption near edge structure (XANES) spectra at the Ce
L3-edge of La
1−xCe
xCoO
3 were analyzed by the conventional superposition method, as in some earlier works [
31,
32,
33,
34]. We performed the first derivative test to find the extremum points. The result gave two local maximum points and one minimum point, corresponding to two component peaks and the valley between them. We also determined an inflection point by inspecting the second derivative. In total, three line-shapes were used in a least-squares fit to the XANES data: two Gaussian line-shape functions to describe the localized final states and an arctangent edge jump function to represent the transition into continuum states.
To explain the observed XANES spectra at the La
L3 edge, reported in the next part of this paper, we put forward a comprehensive characterization of La–O bonding in the main lattice phase of three lattice models. First, we reconstructed the crystal electron density directly, based on the results of the Rietveld refinement for the XRD data [
35]. The spatial resolution of the model electron density was 0.042 Å. The model electron density provided us with the topological analysis of the intermolecular interactions between La and O. We then calculated the electron energy distribution from the experimental electron density, which is a straightforward way to recognize the atomic interaction type of La–O in the lattice model of interest. The theory of this approach is described in detail in the work of Tsirelson [
36]. Utilities of the Visualization for Electronic and Structural Analysis (VESTA) software (Koichi Momma, Ibaraki, Japan) [
37] were used for model visualizations and two-dimensional data display.
The EXAFS data at the Co
K-edge were analyzed using the Demeter package [
38]. We performed XAS data processing in the Athena program. The multi-step procedure resulted in the extracted EXAFS oscillation and Fourier transform for each XAS spectrum, which was then fitted in the Artemis program. Forward Fourier transformations of the EXAFS data were performed in the
range of 3.3–16 Å
−1 using the Kaiser-Bessel window.
We conducted an unconventional approach [
39,
40] to include a mixture of two scattering phases in each sample. Theoretical scattering paths, as listed in
Table 1, were generated with the structural parameters obtained from the Rietveld analyses for the XRD data. For the sample with
x = 0, we assumed a mixture of a rhombohedral main lattice and a cubic Co
3O
4-like phase. For both the
x = 0.2 and
x = 0.4 samples, we considered a combination of a cubic main lattice and a cubic Co
3O
4-like phase. To interpret the introduced structure of Co
3O
4 in a manner compatible with the Demeter program, we transformed the non-standard setting
Fd-3m:1 [
28] into the standard
Fd-3m using a shift vector of (1/8, 1/8, 1/8). The list of potentials was complemented with a new potential index and the atomic number for Ce. Changes of La tags to be Ce in the list of atom coordinates were also done in addition to appropriate adjustment of the corresponding potential index before running the FEFF calculation. We aimed to deal with the information-limited problem to fit for a
range of 1–4 Å. The relation
suggested approximately 24 independent parameters. Normal “strict degeneracy” was used for the FEFF calculation for the main scattering phase, but the so-called “fuzzy degeneracy” [
41] with a margin of 0.03 Å was applied for the aggregate FEFF calculation over two sites of the Co
3O
4-like scattering phase. The number of scattering paths was therefore reduced substantially to only three single scattering paths (SS), as listed in
Table 2 and
Table 3 for the secondary scattering phase. We also included two multiple scattering paths in the model for the main lattice, that is, a three-legged path @O1.1Co1.1@ and a four-legged path @O1.1Co1.1O1.1@. In a cubic main phase, these forward scattering paths are perfectly focused and do not put in new free parameters to a fit [
39]. In a rhombohedral main phase, these paths are partially focused. Since the angle with the vertex at Co is only 8.5° [
39], as determined from the result of the Rietveld refinement, both paths can be treated as the same as the single-scattering path @Co1.1@. However, we found that the quality of the fit could be statistically improved by strictly defining the
of the path @O1.1Co1.1@ as
, as shown in
Table 2. We used an expansion factor
α and the built-in
Reff value to describe the isotropic change of all paths of any cubic phase. The variable for
of the Co
3O
4-like scattering phase is expressed as α
Reff in
Table 2. In
Table 3, the settings α
1Reff and α
2Reff account for the change in distance of all paths of the main lattice and the secondary phase, respectively.
Regarding all the above-mentioned details, we parametrized 14 variables for
,
, and
for the case
x = 0 and 10 variables for the cases
x = 0.2 and
x = 0.4, as displayed in
Table 2 and
Table 3, respectively. Additionally, there was one more variable not shown in these tables, which accounts for the amplitude term
for each case of
x. The amplitudes of the main scattering phase and the secondary phase correlate by the formula
. We implemented fits to the data simultaneously with multiple
k weightings (i.e.,
k to
k3). Phase correction for the radial distance was made based on the Co–O pair of the main scattering phase.
4. Results and Discussion
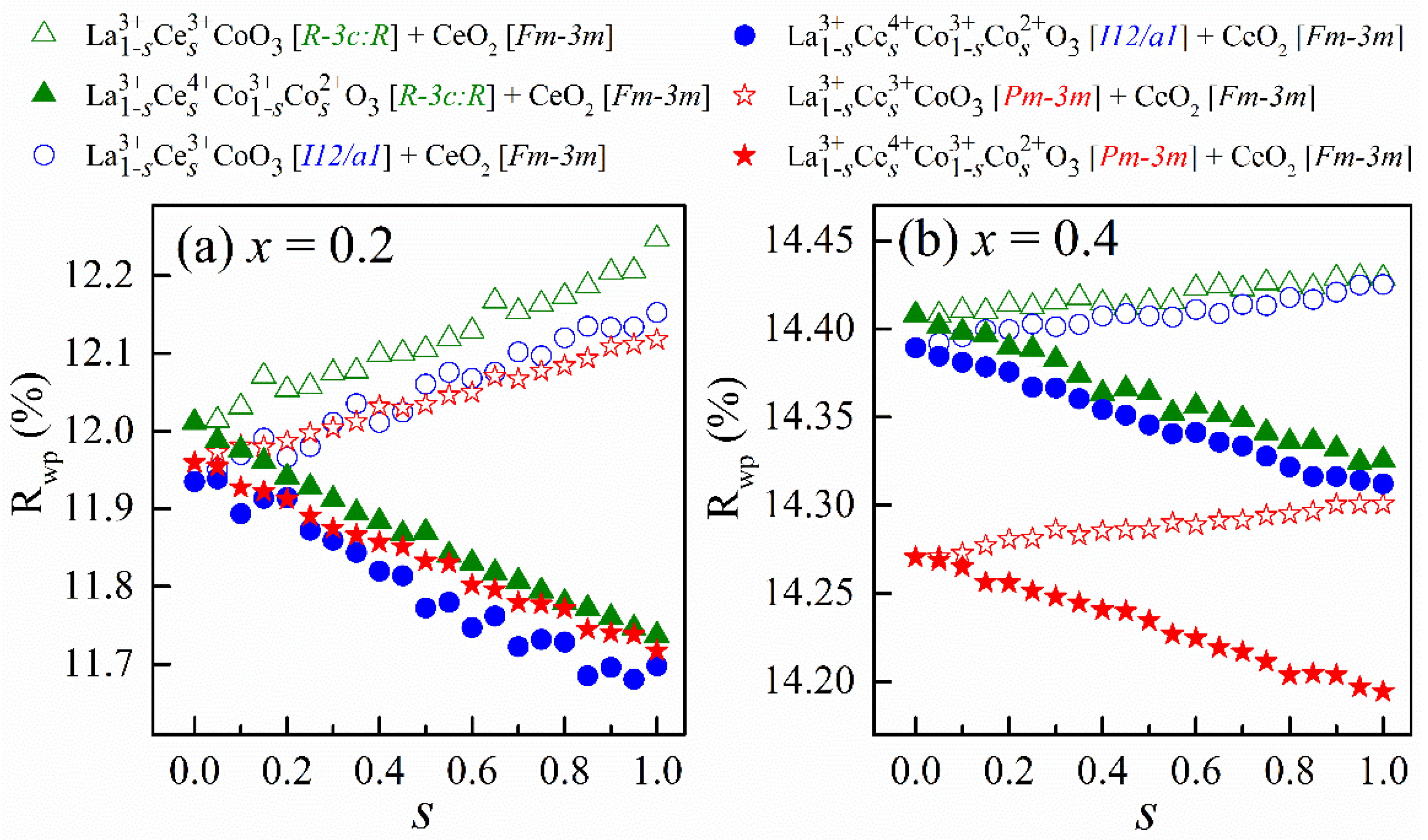
Figure 1 shows
Rwp as a function of
s for 252 fits to the experimental XRD data of La
1−xCe
xCoO
3. It is worth repeating that
s accounts for the atomic percent of Ce in La
1−sCe
sCoO
3, the main lattice. Such use of
s is to conveniently tackle the question as to whether Ce is present and to what extent in the host lattice. The inquiry emerges from a prior admission [
20] for an already formed secondary phase, CeO
2, if the nominal composition of Ce is higher than 0.03. We find that the two-phase model results in statistically better refinement than the single-phase model. All the resultant
Rwp are lower than 15%, which meets the requirement for an adequate Rietveld refinement. Both cases of
x reveal that when combining Ce
4+ and Co
2+ as substitutes for La
3+, the
Rwp are lower than those where the dopants are Ce
3+, highlighting the importance of the charge compensation by Co
2+. However, since the
Rwp changes linearly and nearly monotonically with
s, the absolute extent of Ce insertion into the host lattice remains inconclusive. Further, for
x = 0.2, the
Rwp regarding each form of dopant are comparable among the three space groups of the main lattice. For
x = 0.4, respecting space groups of the main lattice, the
Rwp for cubic
Pm-3m are lower than those for rhombohedral
R-3c: R and monoclinic
I12/a1, which holds for both forms of dopants under consideration.
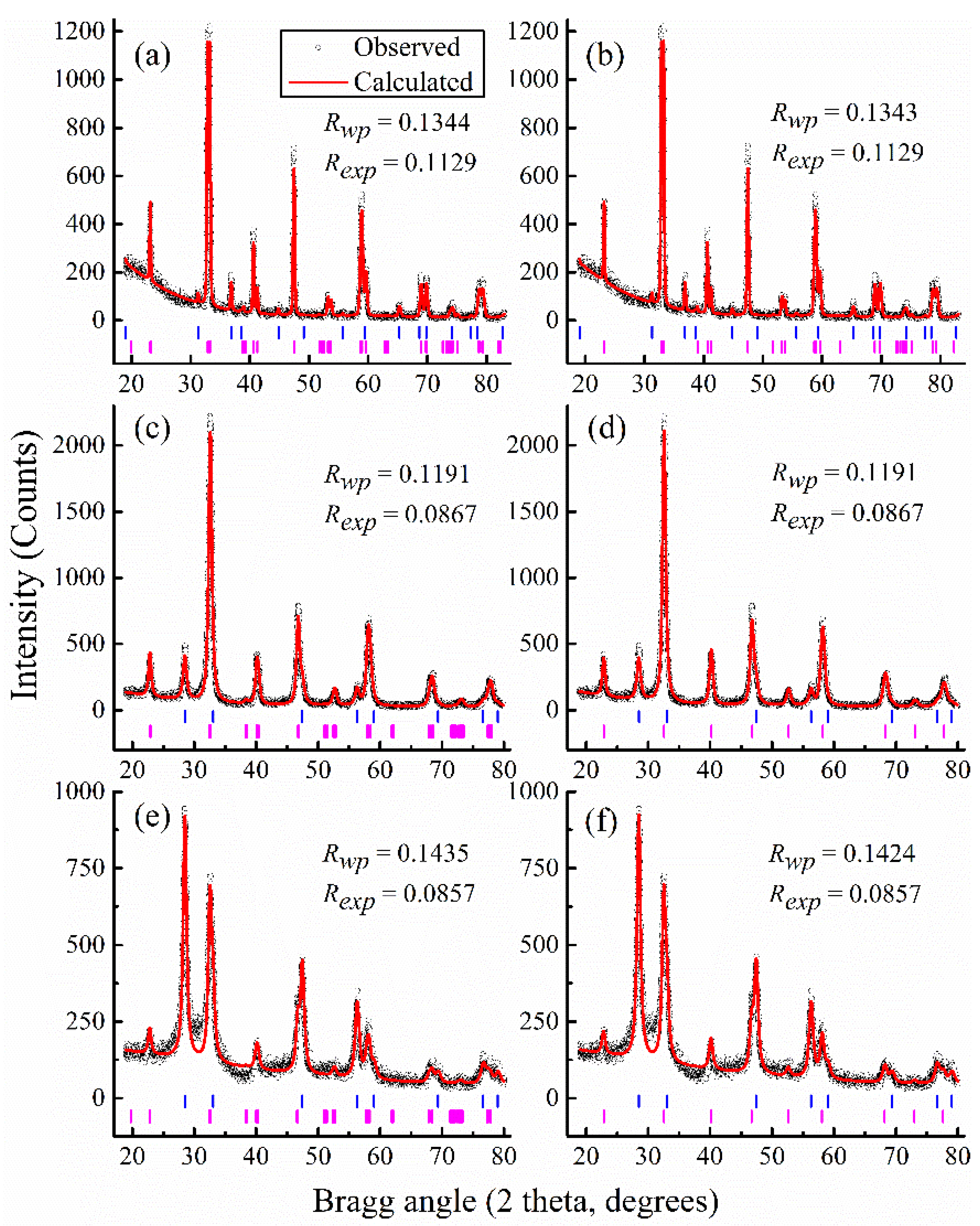
Figure 2 displays some selected Rietveld refined XRD patterns along with the corresponding Bragg positions. It is noteworthy that the cubic space group
Pm-3m applied for the main lattice does not result in a successful fit for
x = 0 and so is not reported.
Figure 2a,b is for the case
x = 0, for which the
R-factors are 0.1343 and 0.1344, corresponding to the main lattice as rhombohedral
R-3c: R and monoclinic
I12/a1, respectively. The fact that the two spectra are identical and have equivalent
R-factors is consistent with the indistinguishable XRD patterns [
7] found previously between the two assumed space groups. Due to this similarity, we show only the typical spectra of monoclinic
I12/a1 to compare with the cubic
Pm-3m in
Figure 2c,d for
x = 0.2 and in
Figure 2e,f for
x = 0.4. The graphic analysis of these patterns brings about an agreement with the
Rwp reported in
Figure 1. However, the vertical bars representing positions of the Bragg peaks located at the bottom of each figure indicate the disappearance of many peaks for the monoclinic
I12/a1 compared to the cubic
Pm-3m. These aspects, together with the discussion for
Figure 1, show that the cubic model performs best for the interpretation of the main lattice in the crystals of Ce-doped LCO.
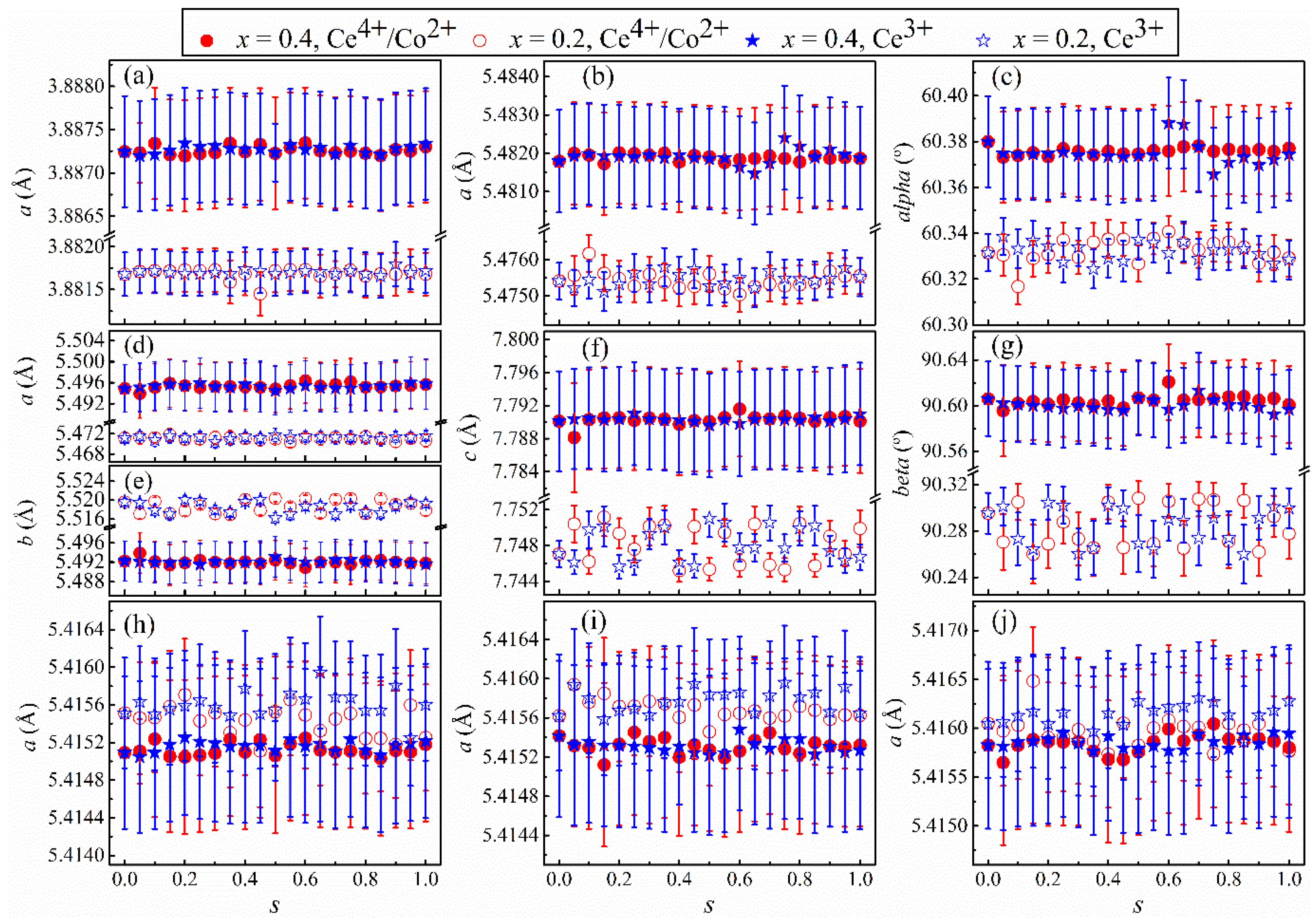
Figure 3 shows the refined lattice parameters from the same fits that resulted in the
Rwp in
Figure 1. The refined parameters as a function of
s for the main lattice in
Figure 3a–g are explicitly different between the two cases of
x, but only within the error bars for the secondary phase in
Figure 3h–j. In
Figure 3a, values for the lattice parameter
a are greater for
x = 0.4 than for
x = 0.2, but those for different forms of dopants in each
x show no explicit difference. This scenario demonstrates that the expansion of the cubic lattice governed by the doping effect dominates the influence of the intrinsic properties of the dopants, such as the atomic sizes. It is clear from
Figure 3b,c that both the lattice length
a and angle
α increase with increasing
x, indicating the simultaneous doping-derived augmentation and tilting of the rhombohedral lattice. Finally, the lattice parameters of the monoclinic main phase in
Figure 3d–g show mixed trends with increasing
x:
a,
b, and
β increase, but
b decreases.
Additionally,
Table 4 provides the weight percent and details of microstructures for both phases involved in each fit. Notably, the crystallite sizes of the Ce-doped LCO are about six times smaller than the pristine LCO, regardless of the models used in the fits. This shrinkage complies with the use of the dopants and the sol-gel method for sample preparation.
Table 5 summarizes the average lattice parameters to give a comparison among different
x.
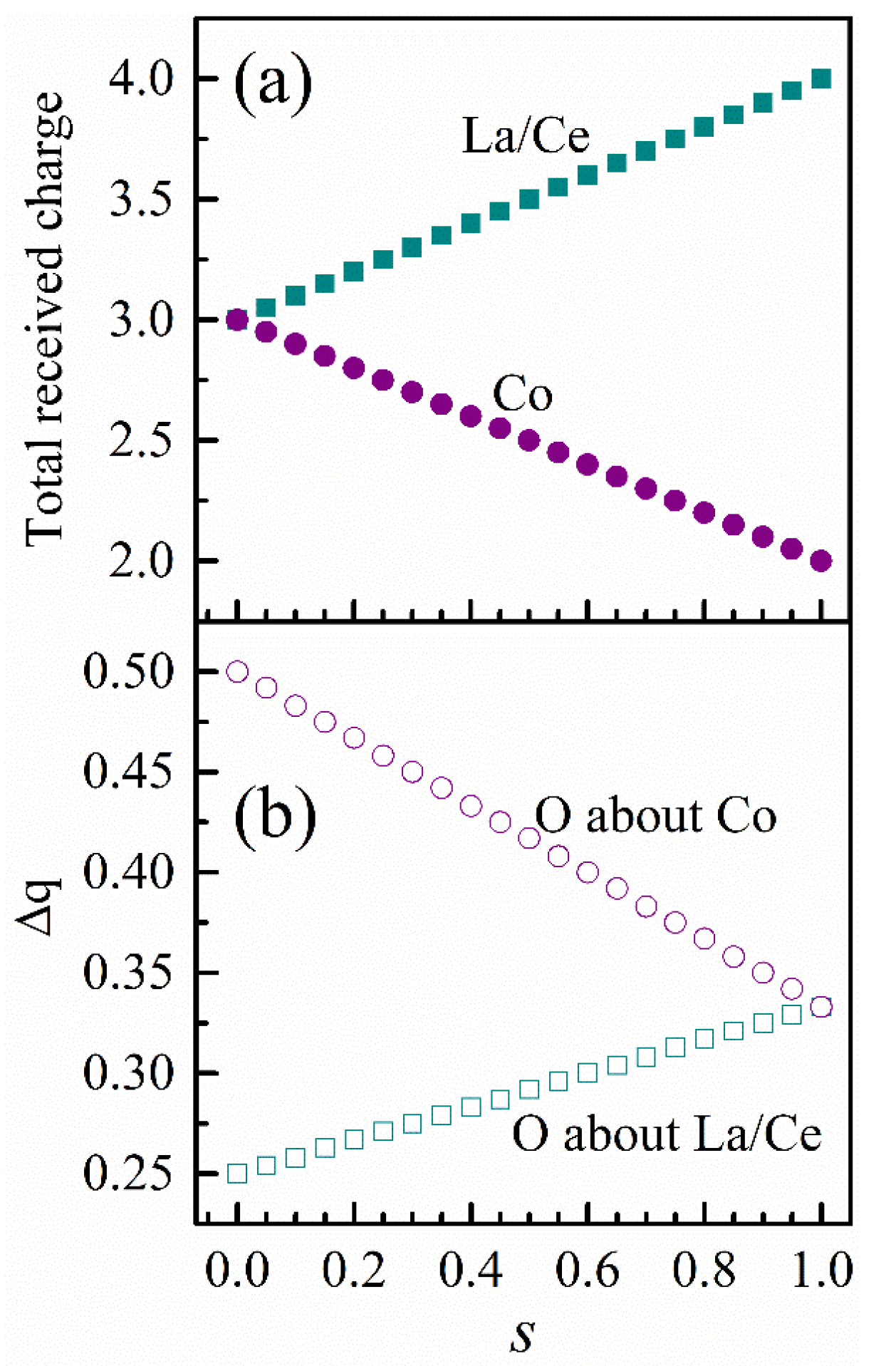
Table 6 tabulates selected bond lengths obtained from the Rietveld refinement and the charge fractions received by each coordination atom, some of which are illustrative in
Figure 4. Noticeably, the Co–O bond length is of a six-fold degeneracy in the rhombohedral main phase, while that in the monoclinic main phase differentiates into three pairs of Co–O bond lengths. This finding is consistent with the fact that the Jahn-Teller distortion is missing in the rhombohedral lattice and exists in the monoclinic lattice.
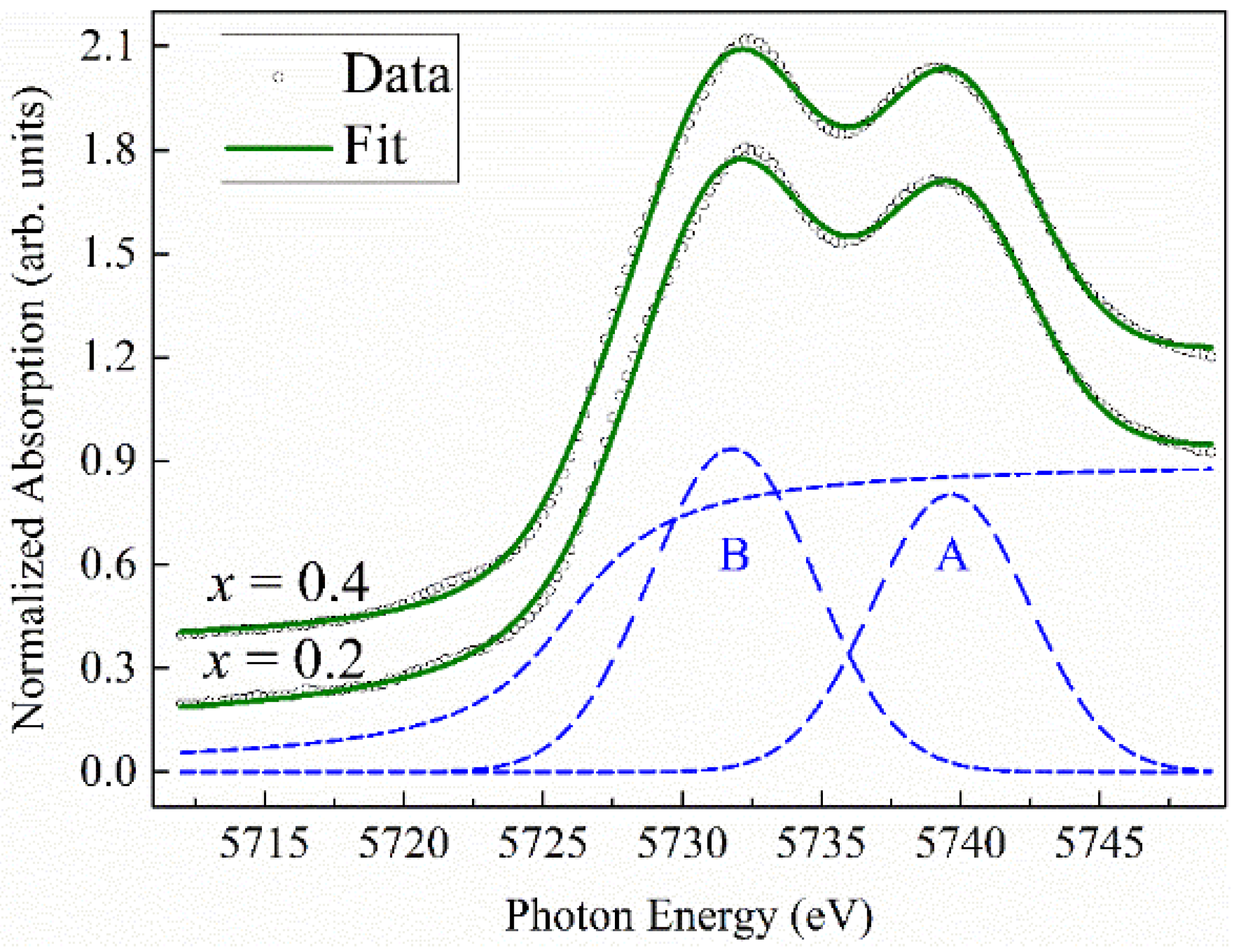
Normalized XANES spectra at the Ce
L3-edge together with the curve fits in
Figure 5 show that Ce of La
1−xCe
xCoO
3 is in a mixed-valence state in both cases of
x studied. It is apparent from the spectra the characteristic two-peak structure, the physical origin of which is elucidative in the work of Kotani and colleagues [
42]. In the 2
p → 5
d photo-absorption, a Ce 2
p core electron absorbs an incident photon and is excited to the Ce 5
d conduction band, leaving behind a core-hole denoted as 2
p3/2. The initial state of the Ce is a superposed state of
[
42,
43], where
c0 and
c1 are real coefficients, and
Lindicates a ligand hole resulting from the O 2
p → Ce 4
f charge transfer. The final state is a consequence of the interplay of three interactions: the Coulomb attraction between the photo-excited 5
d electron (5
d*) and the core-hole, the Coulomb repulsion between the 4
f electron and 5
d*, and the core-hole attractive potential acting on the 4
f electron. The roles of these interactions are to localize the 5
d* near the core hole site, split the Ce 5
d final state into 2
p3/24
f05
d* and 2
p3/24
f1L5
d* configurations, and bring about the 4
f electron relaxation in the final state, respectively. Consistent with this description, we decompose each XANES spectrum into a combination of two Gaussian peaks, describing the 5
d final states and an arctangent edge jump simulating the transition into continuum states. Dashed lines in
Figure 5 illustrate the deconvolution procedure. Peak A represents the Ce tetravalent state with the configuration 2
p3/24
f05
d*. Peak B depicts the 2
p3/24
f1L5
d* state. Using weighted ratio of the areas under peak A (A
4+) and peak B (A
3+), we estimate from the formula 3 + (A
4+/(A
3+ + A
4+)) the Ce spectroscopic valence of 3.41 and 3.45 for
x = 0.2 and
x = 0.4, respectively. These values are smaller than 3.49 [
31] and 3.56 [
34], as previously determined for CeO
2. Hence, the results of spectroscopic valence obtained here might signify at least two crystallographic, distinct Ce sites in the La
1−xCe
xCoO
3 structure.
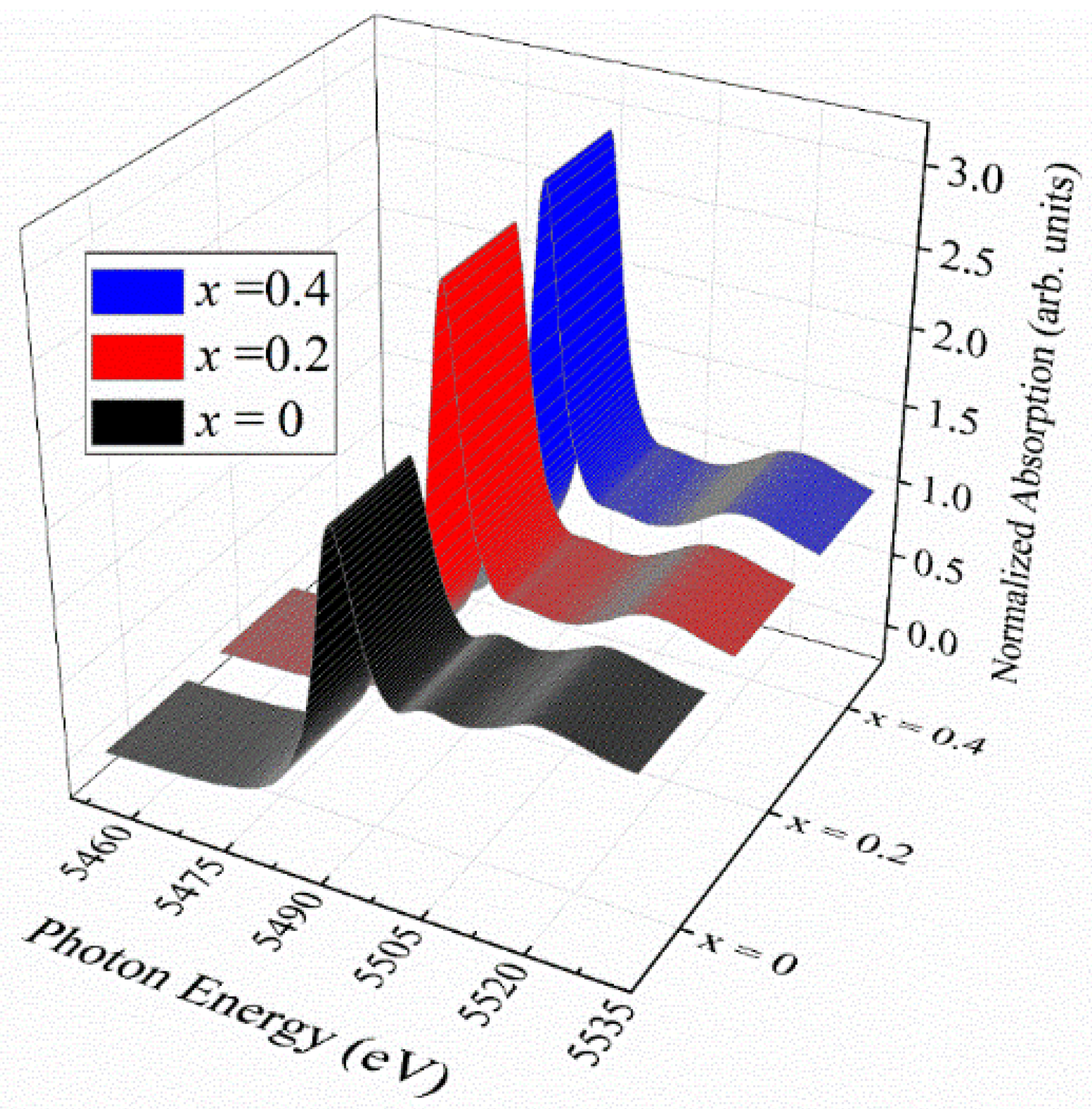
Figure 6 shows XANES data at the La
L3-edge for La
1−xCe
xCoO
3. The spectrum of the pristine LCO reveals a sharp white line and two post-edge bulging features. These structures indicate holes in the La 5
d-band, O 2
p-La 5
d hybridization states, and possibly part of EXASF, respectively. It is clearly visible that the white line becomes enhanced with increasing Ce content, whereas the post-edge structure analogous to the bulge in the middle of the
x = 0 spectrum takes a flat shape in the cases
x = 0.2 and
x = 0.4. These changes establish direct evidence that the La 5
d-band of the pristine LCO is in a partially filled state and eventually turns into a less occupied state when subject to the Ce addition. This finding on the occupancy of the La 5
d-band seems to be unexpected, because the La–O bond is commonly anticipated to be pure ionic, owing to a difference greater than 2.0 between the Pauling’s electronegativity of La (1.1) and that of O (3.44). There is, in fact, a differentiation of the La–O bond length in LCO; La–O bonds with short enough bond lengths have some covalent character, as is revealed in the discussion that follows.
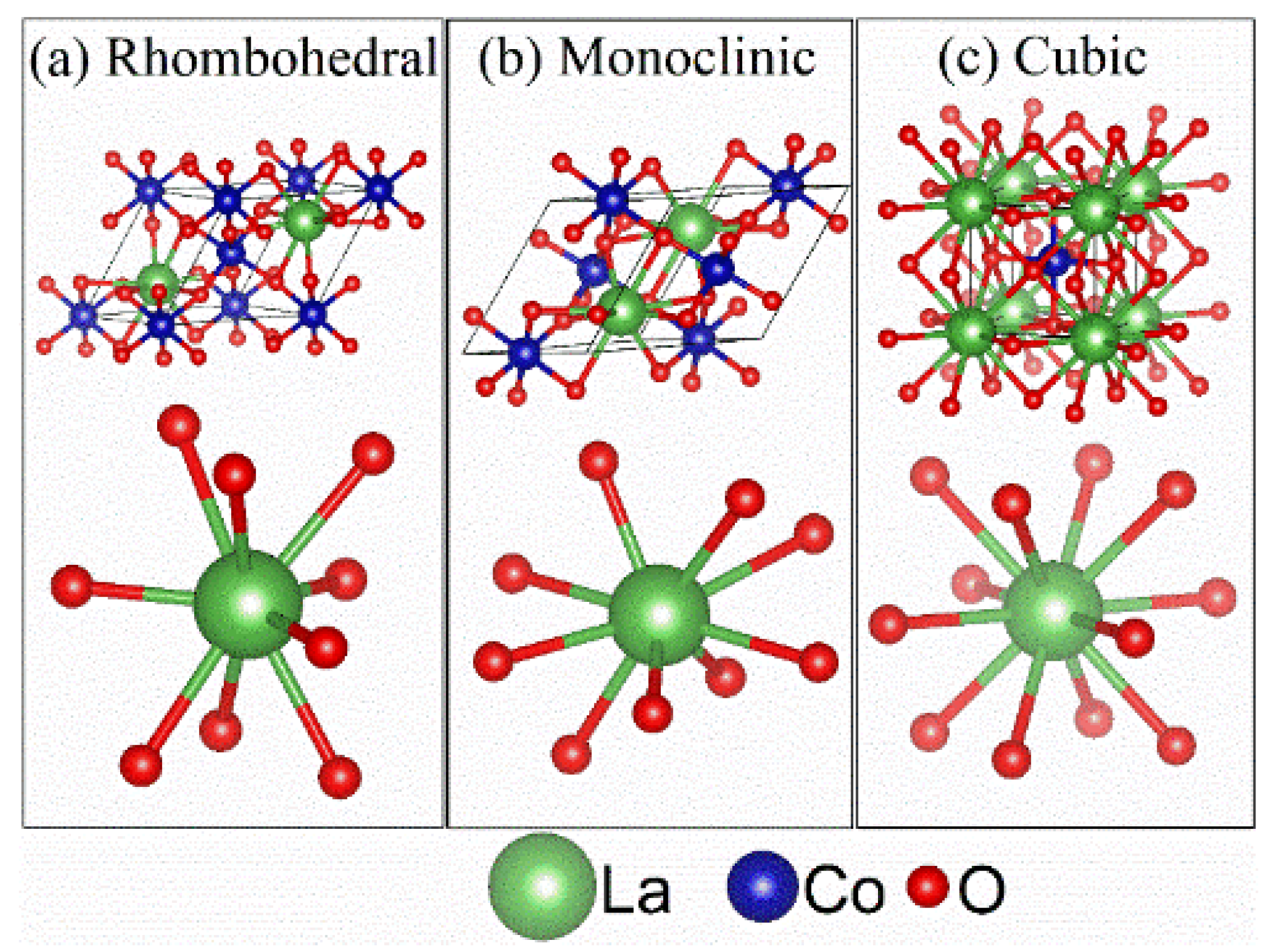
Figure 7 illustrates the local coordination geometry around the La atoms associated with different primitive cells of different lattice models for the main phase of La
1−xCe
xCoO
3. The La atom in either the rhombohedral model or monoclinic model for
x = 0 is surrounded by 9 O atoms, as opposed to 12 in a representative of cubic model for
x = 0.2 and
x = 0.4. This illustration is consistent with the calculation of “effective” coordination number (ECN), using the output from the Rietveld refinement analysis in the present work. The results give the ECN as 8.95 and 8.77 for the rhombohedral model and monoclinic model, respectively, and 12 for the cubic model. More specifically,
Table 6 lists for each lattice model all 12 La–O distances and the fraction of charge received by each coordinating O atom. Regarding the rhombohedral model, there is a threefold degenerate La–O distance of 3.0062 Å, of which each O receives only 0.074 of the charge. In the case of the monoclinic model, there are also three such long La–O distances. They are, however, split into a twofold degenerate distance of 2.991 Å and a sole distance of 3.032 Å. The O atoms corresponding to these distances gain unsubstantial charge fractions as 0.078 and 0.064, respectively. For this reason, the number of effectively coordinate La–O bonds remains ~9 for both the rhombohedral and monoclinic models. In contrast, La is bonded to twelve equivalent O in a 12-coordinate geometry in the cubic model, where all La–O bond lengths are ~2.74 Å. This distinction in the La local structure elucidates the observable changes in the La XANES data in
Figure 6.
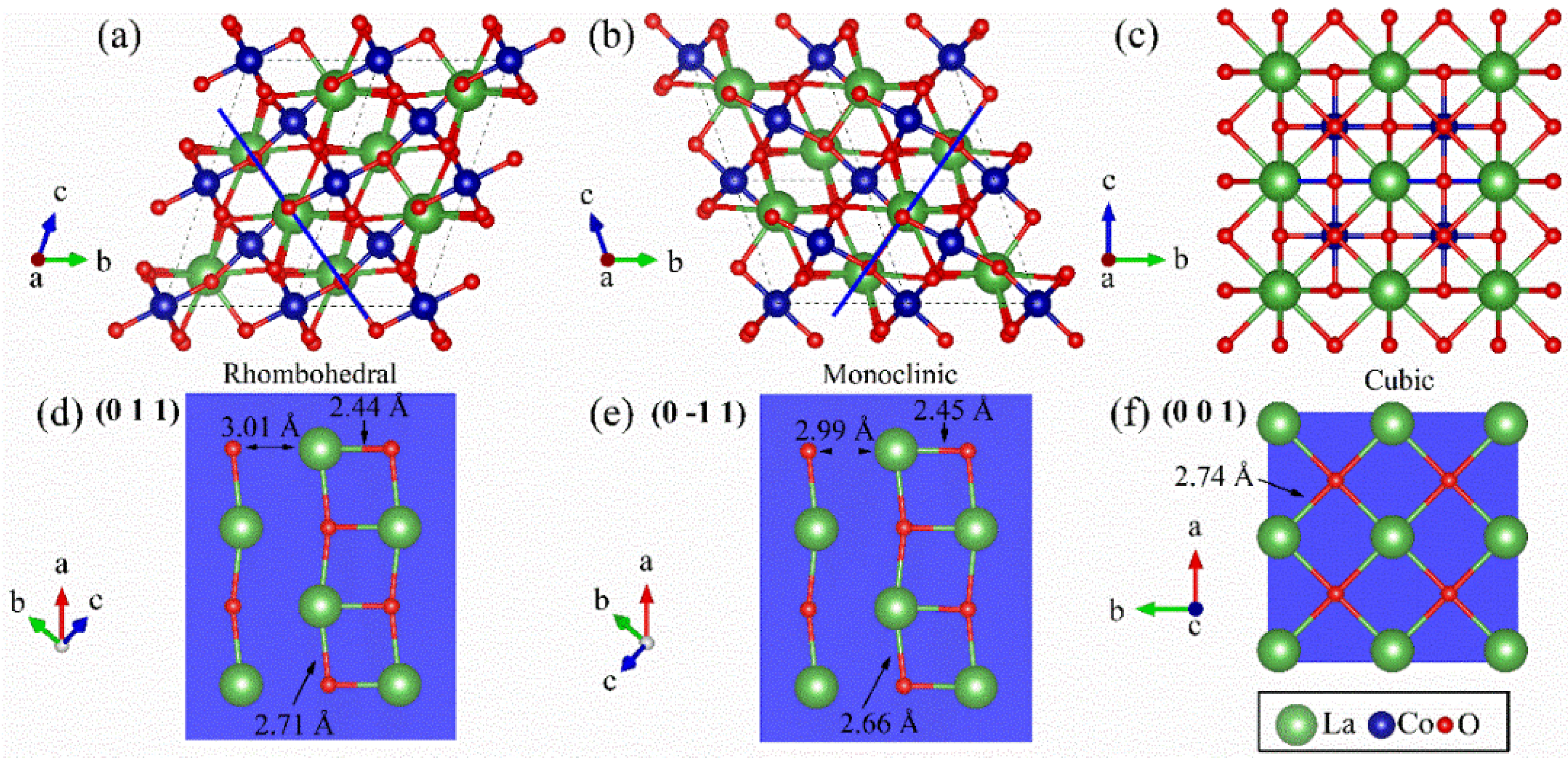
It is necessary to scrutinize the physical nature of interatomic interactions to gain more evidence to support our findings from the La
L3-edge XANES observations. Each of
Figure 8a–c shows a 2 × 2 × 2 supercell of different lattice models for the main phase of La
1−xCe
xCoO
3 along the lattice vector
a. A solid (blue) line therein marks a rhombohedral (011) plane, a monoclinic (0-11) plane, and a cubic (001) plane. In
Figure 8d–f, pictorial details of these planes depict the short (2.4 Å), intermediate (2.7 Å), and long (3.0 Å) La–O distances. These structural descriptions serve as the basis for further considerations of the experimental electron density and the electronic energy distribution shown in
Figure 9a–f, respectively.
Figure 9a explicitly shows an uneven electronic distribution in the rhombohedral (011) plane. Regarding each La atom, which coordinates with 4 O atoms, the interatomic region of a short La–O distance has a higher electron density than that of the three longer La–O distances. Correspondingly,
Figure 9d shows the electronic energy maximally negative in the midway region of the short La–O bond, which unequivocally indicates a shared-type atomic interaction. The observations found here profusely resemble results from inspections for the monoclinic (0-11) plane in
Figure 9b,e. In contrast, the electron density distribution shown in
Figure 9c is uniform for all La–O bonds in the cubic (001) plane. Furthermore, according to the electron energy distribution in
Figure 9f, each La atom in the cubic (001) plane participates in four closed-shell interactions with adjacent O atoms. Overall, these facts infer that some La–O bonding states of the pristine LCO have a covalent bond nature in both rhombohedral and monoclinic lattice models and tend to favor a change towards being nearly pure ionic in cubic Ce-doped LCO.
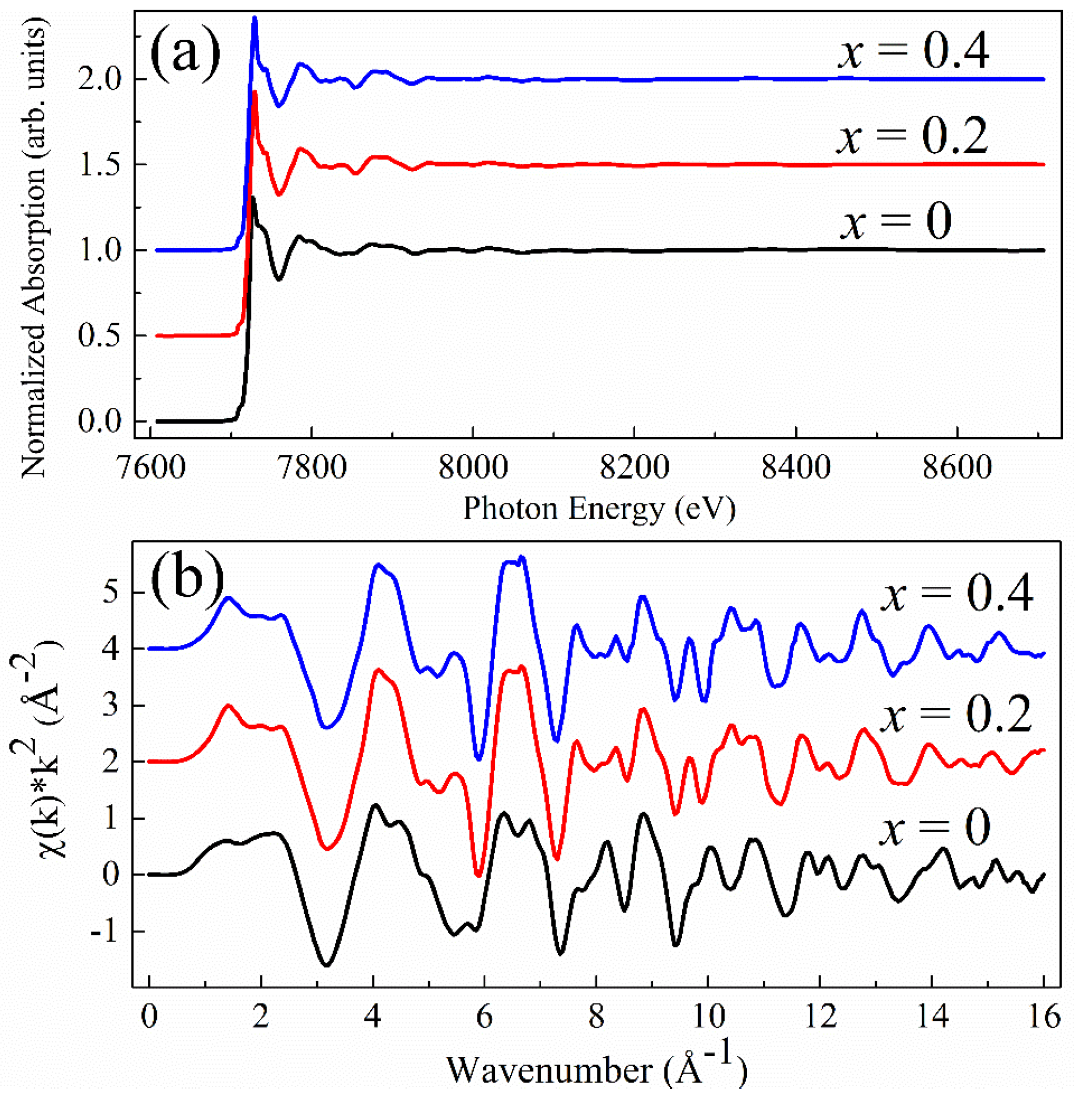
Figure 10 shows the normalized Co
K-edge XAS data measured on the La
1−xCe
xCoO
3 samples and extracted
k2-weighted
data. All the EXAFS fits for
x = 0.2 using different values of
s resulted in intuitively identical curves. The same situation holds for
x = 0.4. Therefore, we show only a representative graph of
s = 0.25 for each case of
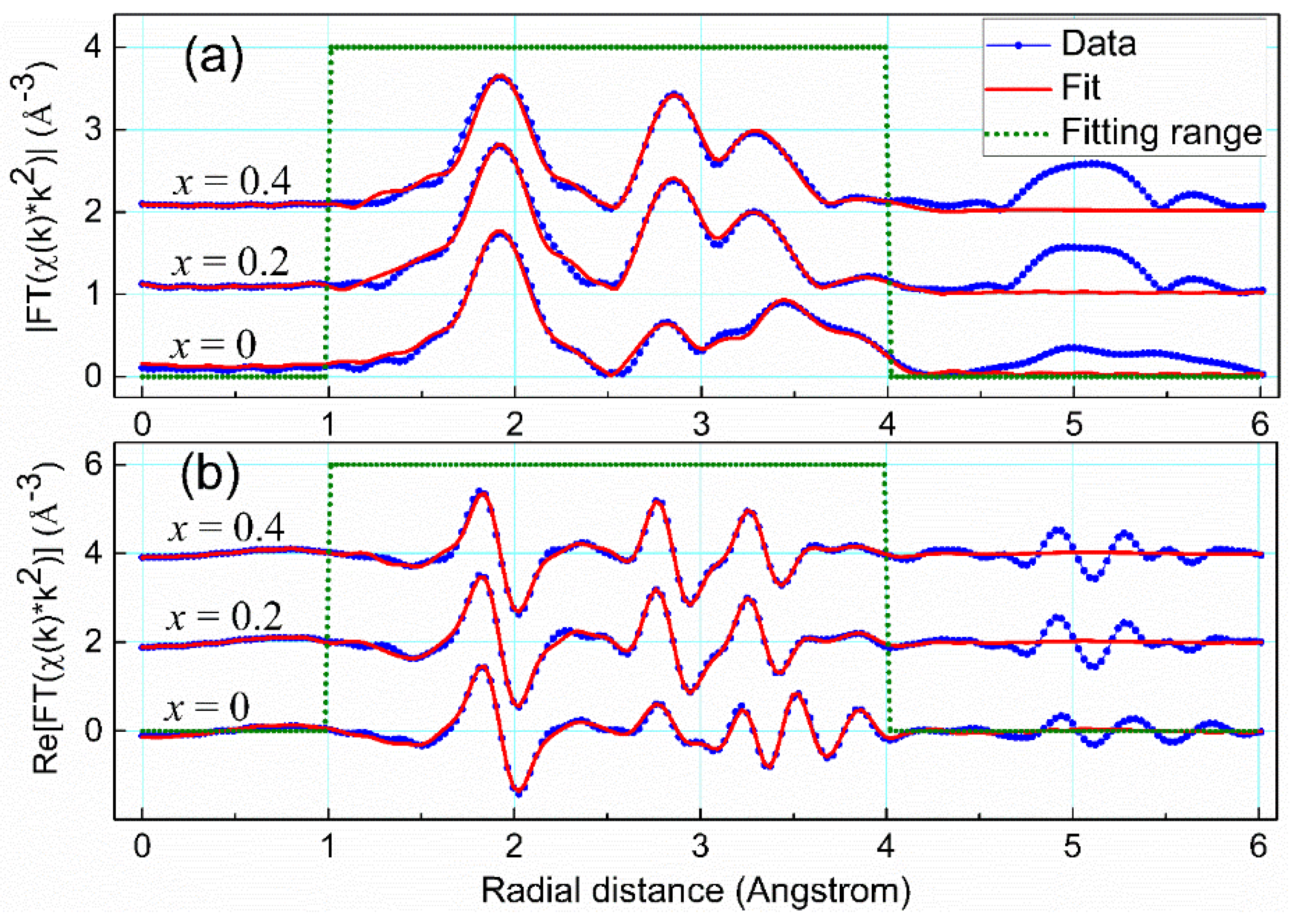
x when comparing the Fourier-transformed EXAFS signals in
Figure 11. It is obvious therein that the theoretical fitting curves match closely with both the magnitude and the real part of the real-space EXAFS data. Moreover, fitted parameters from these fits have adopted reasonable values, as listed in
Table 7,
Table 8 and
Table 9. Minute changes of all the radial distances, except for the value 0.16 Å of the path @La1.1@ in the case
x = 0, justify the Rietveld refinement analysis and the EXAFS models used.
The resulting sigma square terms σ2, accounting for the mean square variation in path lengths, require some discussion. The σ2 value of the path @O1.1@ is smaller in the case of x = 0 than in the cases x = 0.2 and 0.4. Such an attribute depends directly on a difference of one La–O bonding between the rhombohedral lattice and the cubic lattice assumed for the main scattering phase. If LCO crystallizes in the rhombohedral R-3c: R space group, O is bonded in a 5-coordinate geometry to three equivalent La and two equivalent Co atoms. Considering Ce-doped LCO in the cubic Pm-3m symmetry, O is bonded in a distorted linear geometry to four equivalent La and two equivalent Co atoms. To that end, the relative displacement Co → O1.1 in the cubic lattice possesses more disorder than that in the rhombohedral lattice. In addition, the σ2 value of the path @O1.1@ is greater than that of the path @O_SS@ in any case of x. This property also derives from the local coordination geometry. Regarding Co3O4 in the cubic Fd-3m symmetry, O is bonded in a distorted trigonal pyramidal geometry to four Co atoms. Hence, the two scattering phases under consideration differ in both metal-oxygen bonding numbers and atomic coordination types, and the number of metal-oxygen bonding prevails in influencing the disorder of the scattering paths.
In addition, regarding the main scattering phase in each case of
x, the σ
2 value is higher for the path @O1.1@ than other scattering paths, reflecting a reversal dependence of σ
2 on the atomic number of the scattering atom. A similar trend is observable for the Co
3O
4-like phase. In general, σ
2 increases with increasing radial distance. In particular,
Table 7 shows a σ
2 value as high as 0.0296 Å
2 for the scattering path @Co_SS2@, whereas the corresponding values in
Table 8 and
Table 9 are 0.0052 Å
2 and 0.0053 Å
2, respectively. This seems to indicate that the Co
3O
4-like phase is only well-ordered in a shorter range for
x = 0 than for
x = 0.2 and
x = 0.4. It is worth reminding that Co
3O
4 crystallites are noticeable from the XRD pattern of the case
x = 0 and missing for
x = 0.2 and
x = 0.4. We thus attribute the difference herein to the degrees of freedom in the fits, i.e., the model with the cubic main lattice has more degrees of freedom than the model with the rhombohedral main lattice.
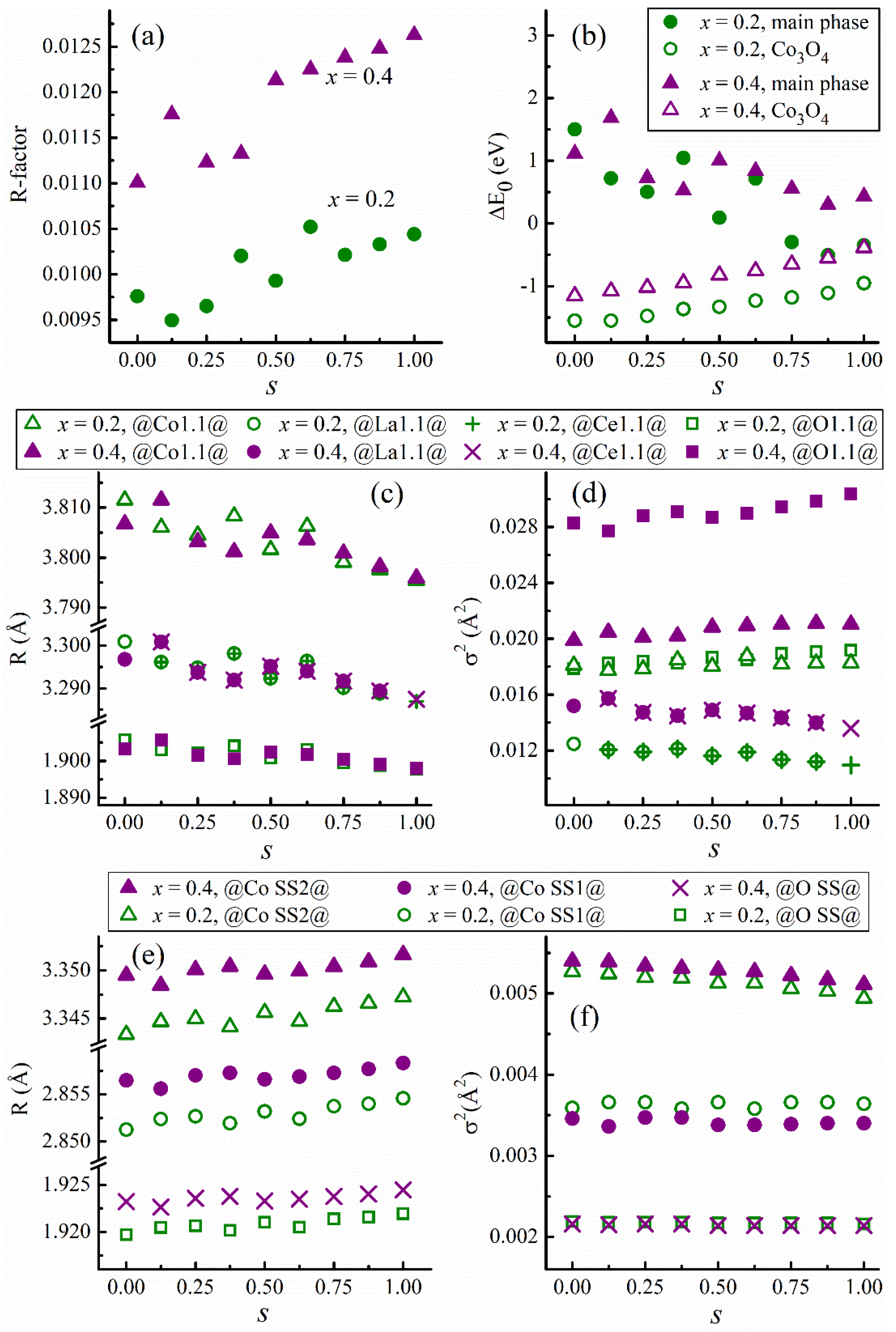
Furthermore,
Figure 12a–f present the full particulars of
R-factor, △E
0, radial distance, and σ
2 as a function of
s for
x = 0.2 and
x = 0.4. All the
R-factors less than 0.02 and the △E
0 much less than 10 eV are strong indications of good EXAFS fits, validating the model of a mixture of two scattering phases. Notably, with increasing
x, the main scattering phase shows subtle changes in the radial distance in
Figure 12c, in contrast to the vividly increased σ
2 values in
Figure 12d. Otherwise, with increasing
x, the secondary scattering phase reveals a distinct change of radial distance in
Figure 12e and an insubstantial variation of σ
2 values in
Figure 12f. We deduce from these observations that the main scattering phase of the
x = 0.4 sample possesses more disorder than that of the
x = 0.2 sample. Additionally, the disorder degree of the secondary scattering phase is similar between the two cases
x = 0.2 and
x = 0.4.


 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}