

Direct Phasing of Coiled-Coil Protein Crystals



Abstract
:1. Introduction
2. Methodology
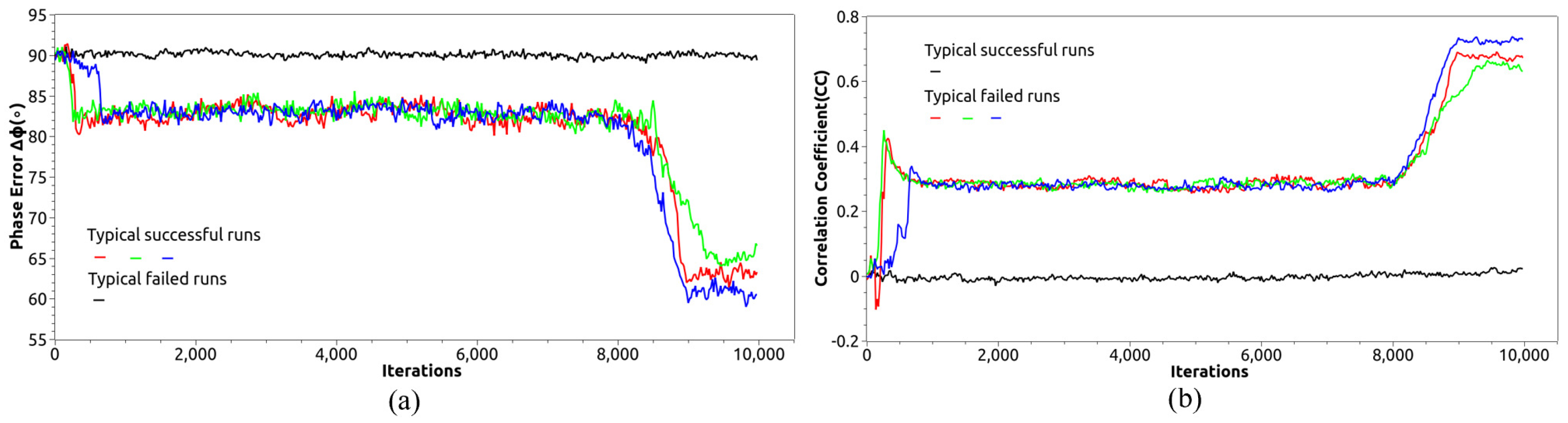
2.1. Direct Phasing Method with Hybrid-Input Output Algorithm
2.2. Direct Phasing with Non-Crystallographic Symmetry Density Averaging
2.3. Direct Phasing of Intermediate- and Low-Resolution Data
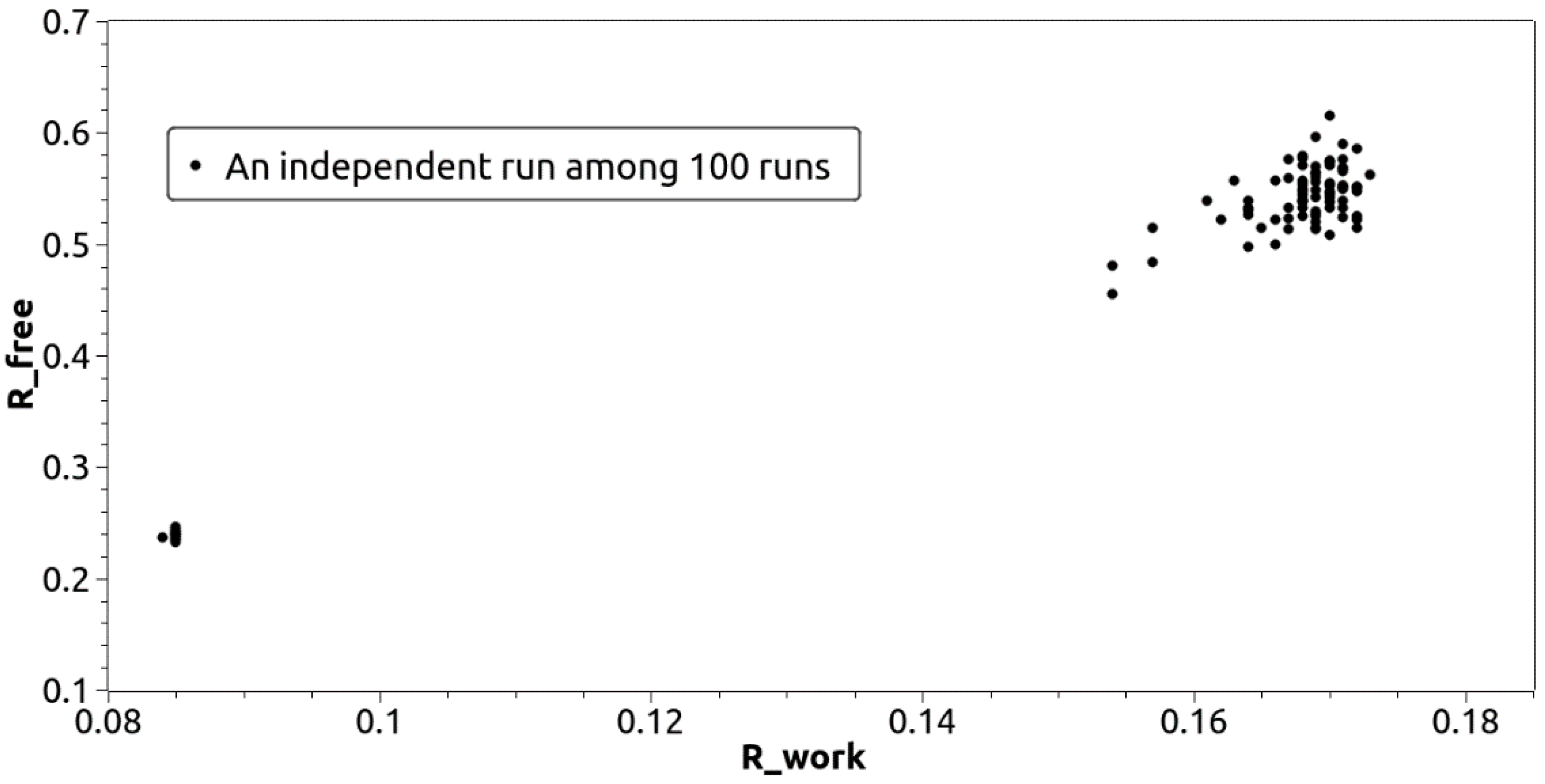
3. Results
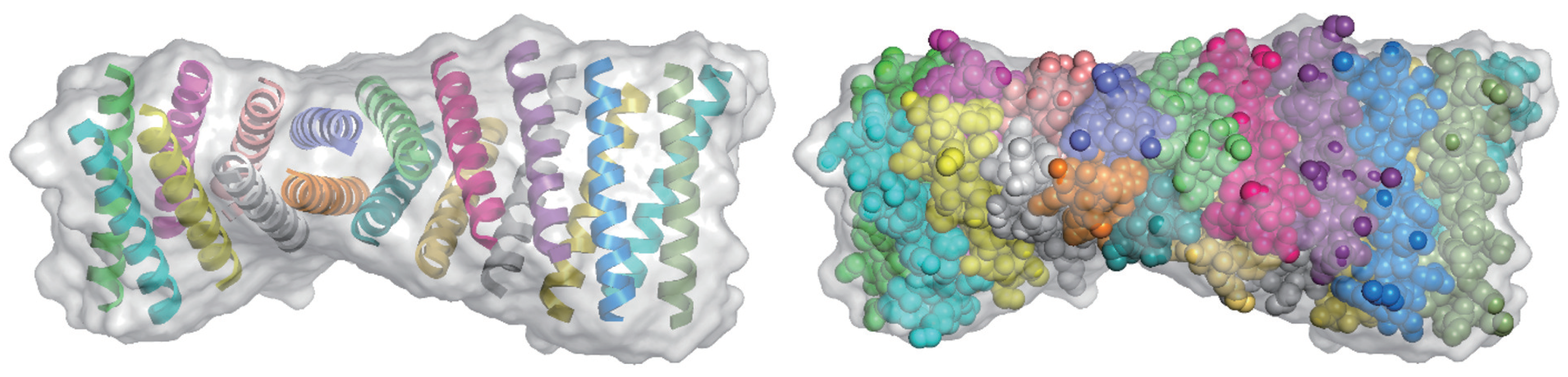
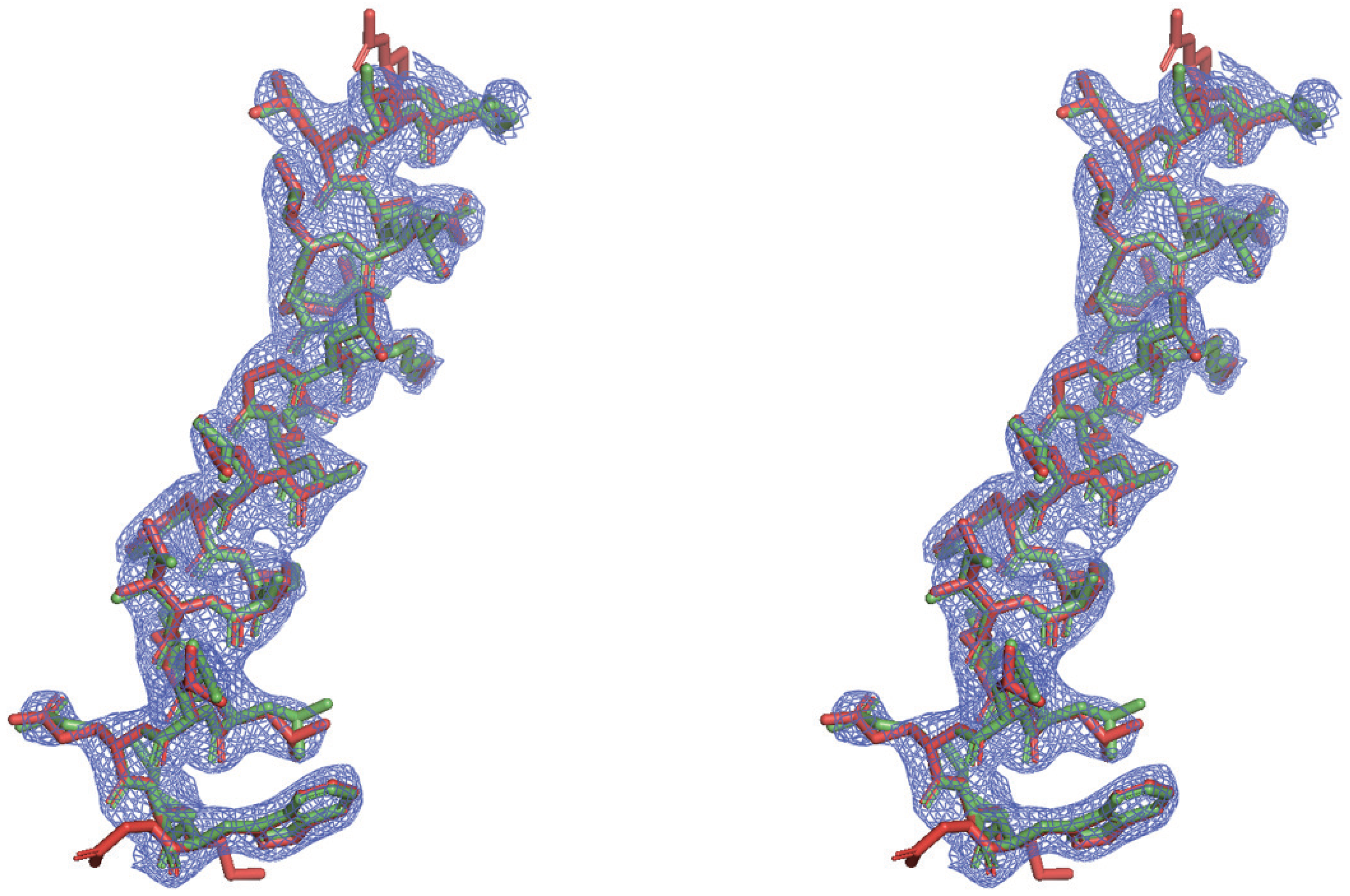
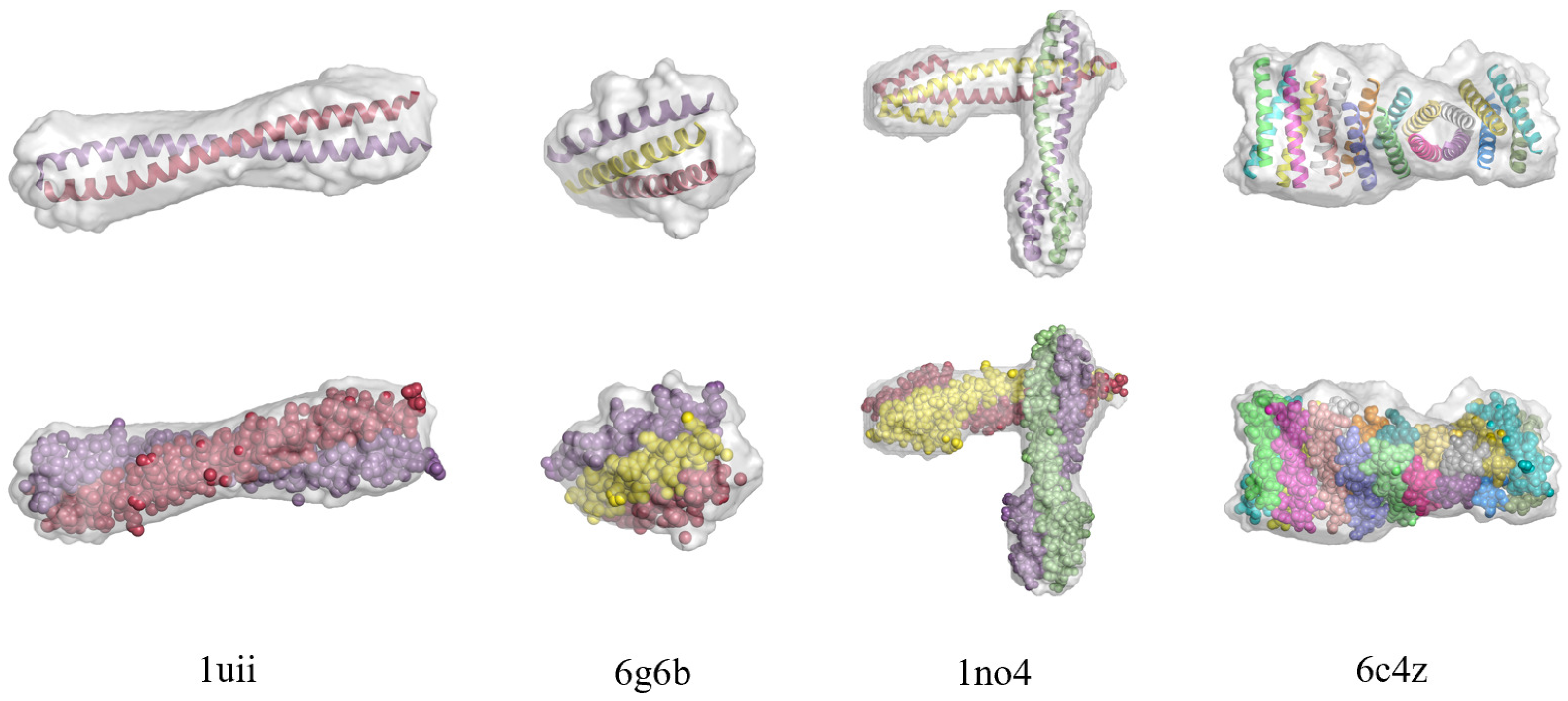
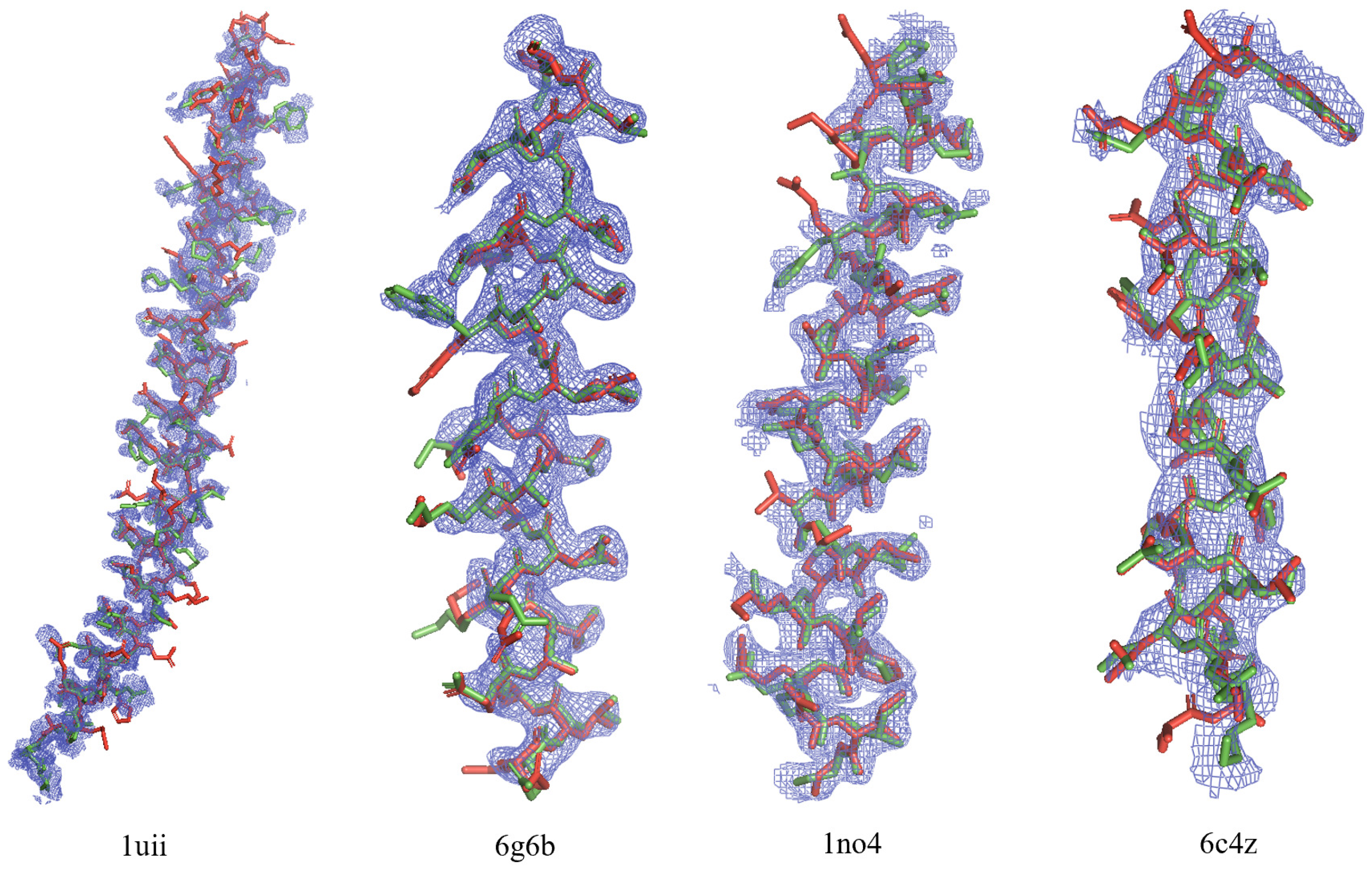
3.1. Direct Phasing of AlphaFold- or MR-Difficult Structures
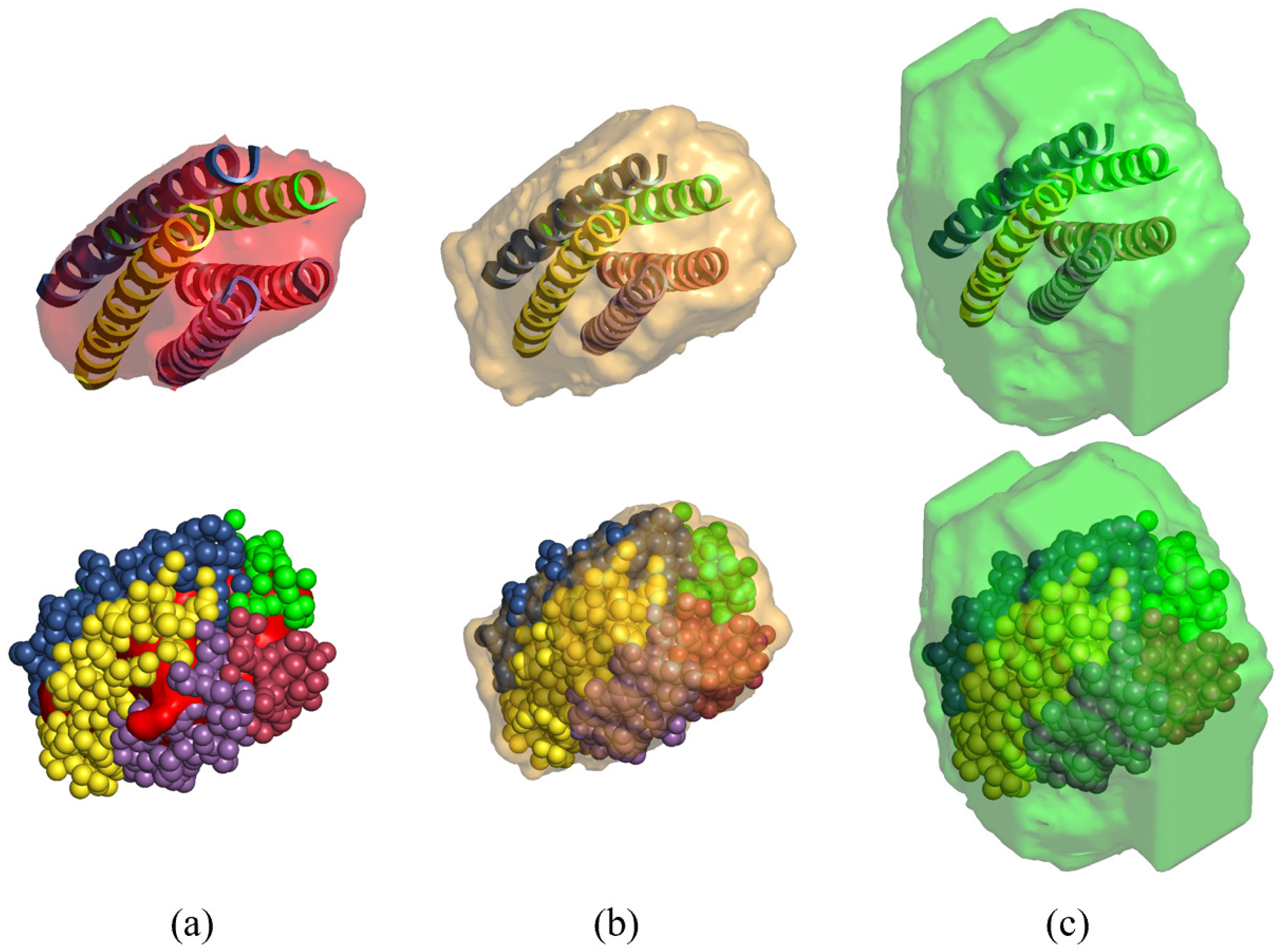
3.2. Direct Phasing of Protein Crystals with Non-Crystallographic Symmetry
3.3. Direct Phasing of Intermediate- and Low-Resolution Diffraction Data
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caballero, I.; Sammito, M.; Millán, C.; Lebedev, A.; Soler, N.; Usón, I. ARCIMBOLDO on coiled coils. Acta Cryst. D 2008, 74, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, G.R.; Wyckoff, H.; Phillips, D.C. A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, W.A.; Smith, J.L.; Phizackerley, R.P.; Merritt, E.A. Crystallographic structure analysis of lamprey hemoglobin from anomalous dispersion of synchrotron radiation. Proteins Struct. Funct. Bioinform. 1988, 4, 77–88. [Google Scholar] [CrossRef]
- Rossmann, M.G.; Blow, D.M. The detection of sub-units within the crystallographic asymmetric unit. Acta Cryst. 1962, 15, 24–31. [Google Scholar] [CrossRef]
- Gillingham, A.K.; Munro, S. Long coiled-coil proteins and membrane traffic. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2003, 1641, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Sayre, D. The squaring method: A new method for phase determination. Acta Cryst. 1952, 5, 60. [Google Scholar] [CrossRef]
- Hauptman, H. A minimal principle in X-ray crystallography: Starting in a small way. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1993, 442, 3–12. [Google Scholar] [CrossRef]
- Zhang, K.Y.J.; Main, P. The use of Sayre’s equation with solvent flattening and histogram matching for phase extension and refinement of protein structures. Acta Cryst. A 1990, 46, 377–381. [Google Scholar] [CrossRef]
- Millane, R.P. Phase retrieval in crystallography and optics. J. Opt. Soc. Am. 1990, 7, 394–411. [Google Scholar] [CrossRef]
- Elser, V. Phase retrieval by iterated projections. JOSA A 2003, 20, 40–55. [Google Scholar] [CrossRef]
- Liu, Z.C.; Xu, R.; Dong, Y.H. Phase retrieval in protein crystallography. Acta Cryst. A 2012, 68, 256–265. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Su, W.-P. Direct phasing of protein crystals with high solvent content. Acta Cryst. A 2015, 71, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Fienup, J.R. Reconstruction of an object from the modulus of its Fourier transform. Opt. Lett. 1978, 3, 27–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fienup, J.R. Phase retrieval algorithms: A comparison. Appl. Opt. 1982, 21, 2758–2769. [Google Scholar] [CrossRef] [Green Version]
- Fienup, J.R. Phase retrieval algorithms: A personal tour. Appl. Opt. 2013, 21, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Millane, R.P.; Stroud, W.J. Reconstructing symmetric images from their undersampled Fourier intensities. J. Opt. Soc. Am. A 1997, 14, 568–579. [Google Scholar] [CrossRef]
- Elser, V. Solution of the crystallographic phase problem by iterated projections. Acta Cryst. A 2003, 59, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, S.; He, H.; Chapman, H.N.; Hau-Riege, S.P.; Noy, A.; Howells, M.R.; Weierstall, U.; Spence, J.C.H. X-ray image reconstruction from a diffraction pattern alone. Phys. Rev. B 2003, 68, 140101. [Google Scholar] [CrossRef] [Green Version]
- Millane, R.P.; Lo, V.L. Iterative projection algorithms in protein crystallography. I. Theory. Acta Cryst. A 2013, 69, 517–527. [Google Scholar] [CrossRef]
- Lo, V.L.; Kingston, R.L.; Millane, R.P. Iterative projection algorithms in protein crystallography. II. Application. Acta Cryst. A 2015, 71, 451–459. [Google Scholar] [CrossRef]
- He, H.; Jiang, M.; Su, W.P. Direct Phasing of Protein Crystals with Non-Crystallographic Symmetry. Crystals 2019, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Kingston, R.L.; Millane, R.P. A general method for directly phasing diffraction data from high-solvent-content protein crystals. IUCrJ 2022, 9, 648–665. [Google Scholar] [CrossRef] [PubMed]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Huang, H.; Yang, J.; Kratochvil, H.T.; Lolicato, M.; Liu, Y.; Shu, X.; Liu, L.; DeGrado, W.F. Designed peptides that assemble into cross-α amyloid-like structures. Nat. Chem. Biol. 2018, 14, 1171–1179. [Google Scholar] [CrossRef]
- Miao, J.; Sayer, D.; Chapman, H.N. Phase retrieval from the magnitude of the Fourier transforms of non-periodic objects. J. Opt. Soc. Am. 1998, 15, 1662–1669. [Google Scholar] [CrossRef]
- Fourier TransformFunctions. Intel R Math Kernel Library 11.3 ReferenceManual; Intel Corporation: Santa Clara, CA, USA, 2015; pp. 1911–1962. [Google Scholar]
- Zhang, K.Y.J.; Main, P. Histogram matching as a new density modification technique for phase refinement and extension of protein molecules. Acta Cryst. A 1990, 46, 41–46. [Google Scholar] [CrossRef]
- Wang, B.C. Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol. 1985, 115, 90–112. [Google Scholar] [CrossRef]
- Brünger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef]
- He, H.; Su, W.-P. Improving the convergence rate of a hybrid input-output phasing algorithm by varying the reflection data weight. Acta Cryst. A 2018, 74, 36–43. [Google Scholar] [CrossRef]
- Miao, J.; Sayre, D. On possible extensions of X-ray crystallography through diffraction-pattern oversampling. Acta Cryst. A 2000, 56, 596–605. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Hassabis, D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Baker, D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Koepke, J.; Krammer, E.M.; Klingen, A.R.; Sebban, P.; Ullmann, G.M.; Fritzsch, G. pH modulates the quinone position in the photosynthetic reaction center from Rhodobacter sphaeroides in the neutral and charge separated states. J. Mole. Biol. 2007, 371, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; He, H.; Cheng, Y.; Su, W.P. Resolution dependence of an ab initio phasing method in protein X-ray crystallography. Crystals 2018, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.S.; Oberdorfer, G.; Xu, C.; Pei, X.Y.; Nannenga, B.L.; Rogers, J.M.; Baker, D. High thermodynamic stability of parametrically designed helical bundles. Science 2014, 346, 481–485. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.C. Finding non-crystallographic symmetry in density maps of macromolecular structures. J. Struct. Funct. Genom. 2013, 14, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.; Yuan, P.; Dhar, S.K.; Senga, T.; Takeda, D.; Robinson, H.; Kornbluth, S.; Swaminathan, K.; Dutta, A. A dimerized coiled-coil domain and an adjoining part of geminin interact with two sites on Cdt1 for replication inhibition. Mol. Cell 2004, 15, 245. [Google Scholar] [CrossRef]
- Rhys, G.G.; Wood, C.W.; Lang, E.J.; Mulholland, A.J.; Brady, R.L.; Thomson, A.R.; Woolfson, D.N. Maintaining and breaking symmetry in homomeric coiled-coil assemblies. Nat. Comm. 2004, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Morais, M.C.; Kanamaru, S.; Badasso, M.O.; Koti, J.S.; Owen, B.A.; McMurray, C.T.; Anderson, D.L.; Rossmann, M.G. Bacteriophage/phi29 scaffolding protein gp7 before and after prohead assembly. Nat. Struct. Mol. Biol. 2003, 10(7), 572–576. [Google Scholar] [CrossRef]
- Thomas, F.; Dawson, W.M.; Lang, E.J.; Burton, A.J.; Bartlett, G.J.; Rhys, G.G.; Woolfson, D.N. De novo-designed α-helical barrels as receptors for small molecules. ACS Syn. Biol. 2018, 7, 1808–1816. [Google Scholar] [CrossRef]
- Burton, A.J.; Thomson, A.R.; Dawson, W.M.; Brady, R.L.; Woolfson, D.N. Installing hydrolytic activity into a completely de novo protein framework. Nat. Chem. 2016, 8, 837–844. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
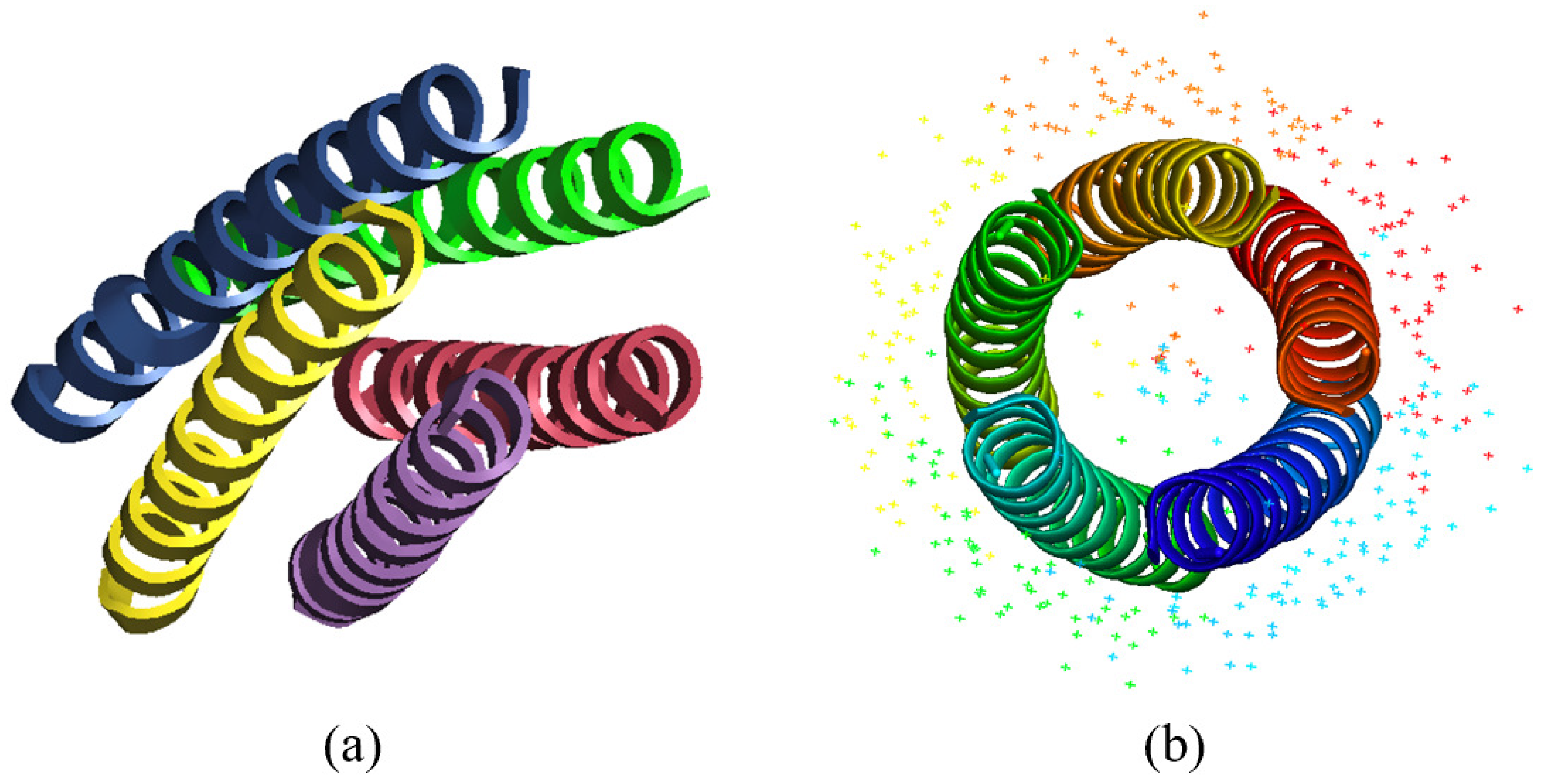{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Space Group | Copied in ASU | Resolution (Å) | Solvent Content (%) | Weight (kDa) | Original Method | Success Rate | Speed of Convergence | Final (°) |
|---|---|---|---|---|---|---|---|---|---|
| 6c4y | P4322 | 18 | 2.5 | 72.31 | 52.53 | MR | 17 | 308 | 39.36 |
| 1uii | P212121 | 2 | 2.0 | 65.30 | 19.53 | MAD | 62 | 260 | 40.29 |
| 6g6b | P4132 | 3 | 2.3 | 74.99 | 9.75 | MR | 35 | 397 | 39.9 |
| 1no4 | P21212 | 4 | 2.2 | 77.7 | 44.66 | MAD | 4 | 3341 | 42.48 |
| 6c4z | P6122 | 18 | 3.3 | 60.38 | 52.53 | MR | 30 | 105 | 58.42 |
| 4uot | C2221 | 5 | 1.69 | 63.9 | 20.68 | MR | 6 | 152 | 29.8 |
| 6eik | P42212 | 7 | 1.52 | 44.89 | 23.97 | MR | 1 | 346 | 42 |
| 5ez8 | P22121 | 7 | 1.95 | 46 | 22.59 | MR | 3 | 114 | 42.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, R.; Su, W.-P.; He, H. Direct Phasing of Coiled-Coil Protein Crystals. Crystals 2022, 12, 1674. https://doi.org/10.3390/cryst12111674
Fu R, Su W-P, He H. Direct Phasing of Coiled-Coil Protein Crystals. Crystals. 2022; 12(11):1674. https://doi.org/10.3390/cryst12111674
Chicago/Turabian StyleFu, Ruijiang, Wu-Pei Su, and Hongxing He. 2022. "Direct Phasing of Coiled-Coil Protein Crystals" Crystals 12, no. 11: 1674. https://doi.org/10.3390/cryst12111674
APA StyleFu, R., Su, W. -P., & He, H. (2022). Direct Phasing of Coiled-Coil Protein Crystals. Crystals, 12(11), 1674. https://doi.org/10.3390/cryst12111674