
Crystalline State Hydrogen Bonding of 2-(2-Hydroxybenzylidene)Thiazolo[3,2-a]Pyrimidines: A Way to Non-Centrosymmetric Crystals

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
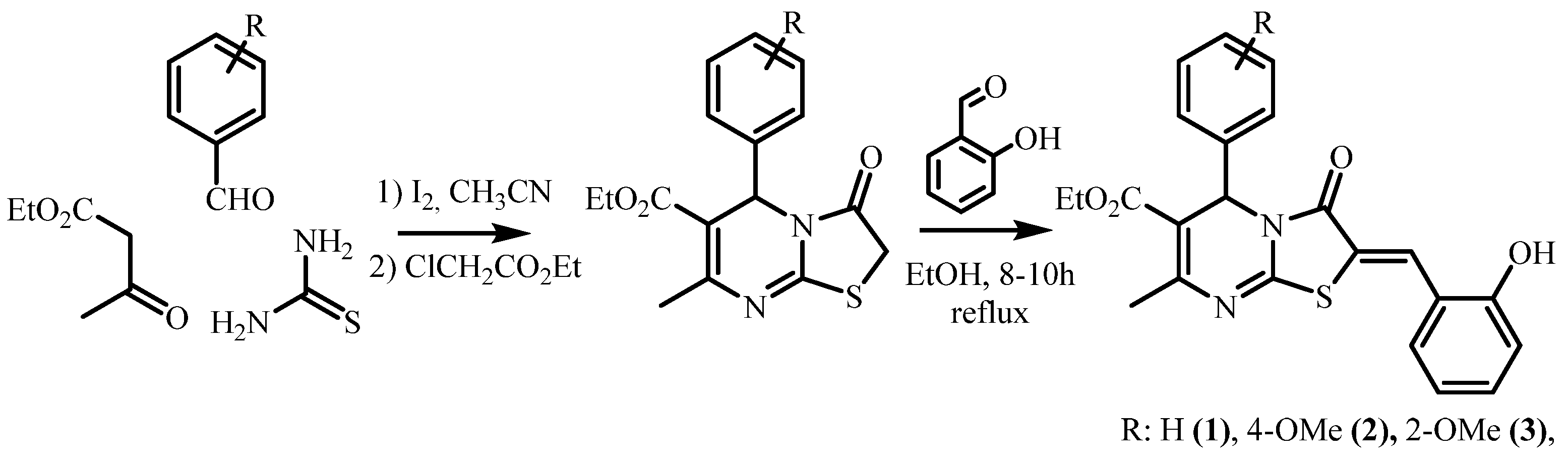
2.1. Synthesis and Crystallization
2.1.1. Synthesis of Ethyl (Z)-2-(2-Hydroxybenzylidene)-7-Methyl-3-Oxo-5-Phenyl-2,3-Dihydro-5H-Thiazolo[3,2-a]Pyrimidine-6-Carboxylate, Compound 1
2.1.2. Synthesis of Ethyl (Z)-2-(2-Hydroxybenzylidene)-5-(4-Methoxyphenyl)-7-Methyl-3-Oxo-2,3-Dihydro-5H-Thiazolo[3,2-a]Pyrimidine-6-Carboxylate, Compound 2
2.1.3. Synthesis of Ethyl (Z)-2-(2-Hydroxybenzylidene)-5-(2-Methoxyphenyl)-7-Methyl-3-Oxo-2,3-Dihydro-5H-Thiazolo[3,2-a]Pyrimidine-6-Carboxylate, Compound 3
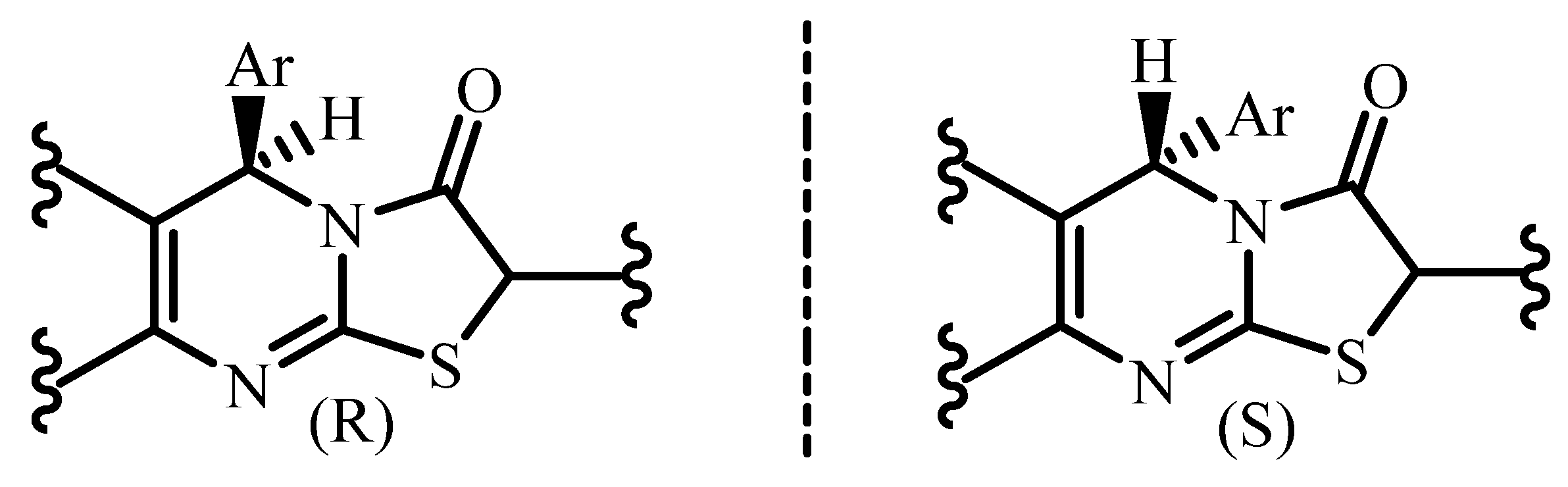
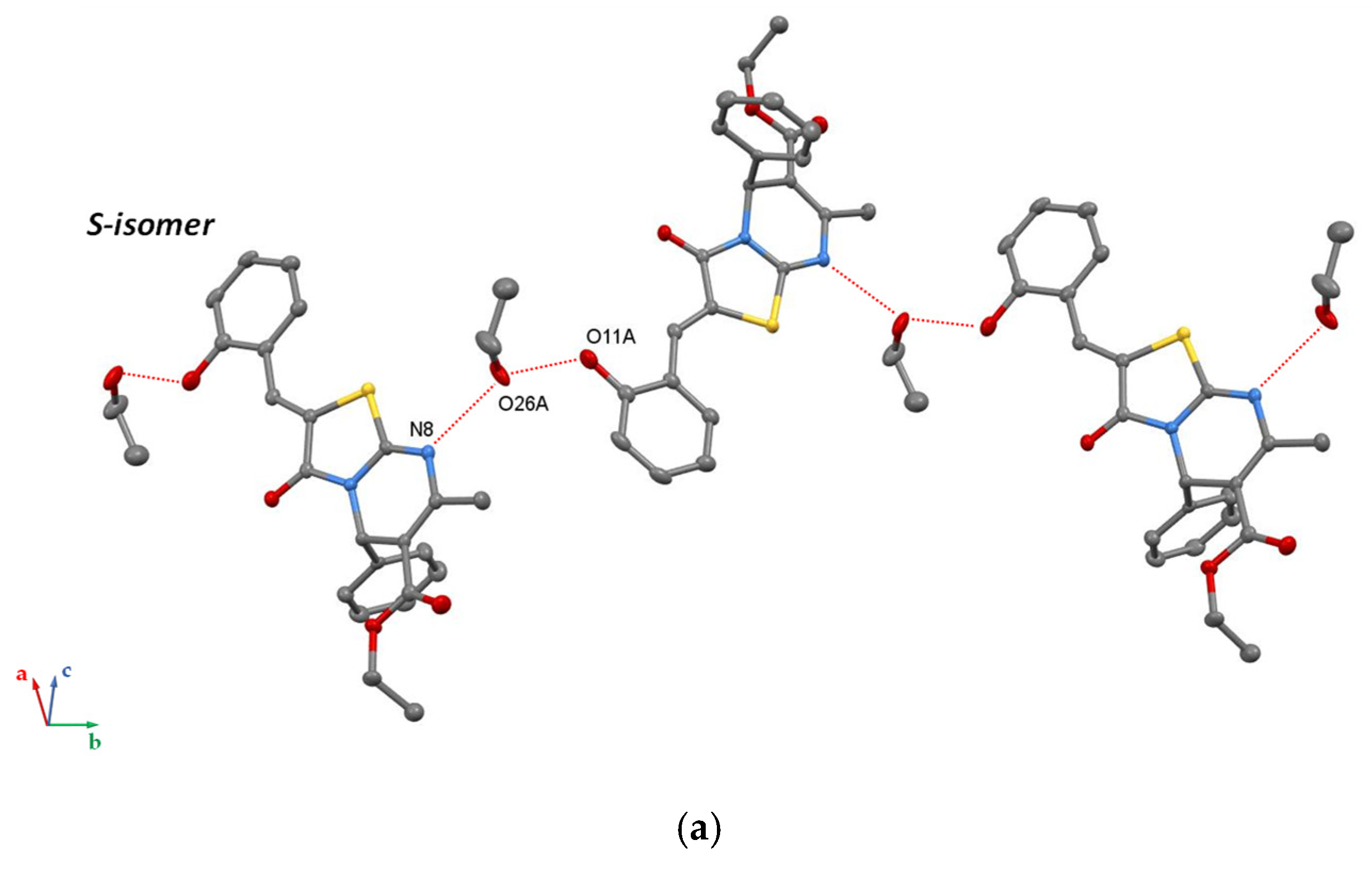
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beaglehole, R.; Bonita, R.; Horton, R.; Ezzati, M.; Bhala, N.; Amuyunzu-Nyamongo, M.; Mwatsama, M.; Reddy, K.S. Measuring progress on NCDs: One goal and five targets. Lancet 2012, 380, 1283–1285. [Google Scholar] [CrossRef]
- Herbstein, F.H. Crystalline Molecular Complexes and Compounds: Structures and Principles; Oxford University Press: Oxford, UK, 2005; Volume 18. [Google Scholar]
- Mayer, T.U.; Kapoor, T.M.; Haggarty, S.J.; King, R.W.; Schreiber, S.L.; Mitchison, T.J. Smart molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999, 286, 971–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshari, A.K.; Singh, A.K.; Saha, S. Bridgehead nitrogen thiazolo[3,2-a]pyrimidine: A privileged structural framework in drug discovery. Mini Rev. Med. Chem. 2017, 17, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jin, Y.; Fu, W.; Xiao, S.; Feng, C.; Fang, B.; Gu, Y.; Li, C.; Zhao, Y.; Liu, Z.; et al. Design, Synthesis, and Structure–Activity Relationship Analysis of Thiazolo[3,2-a]pyrimidine Derivatives with Anti-inflammatory Activity in Acute Lung Injury. ChemMedChem 2017, 12, 1022–1032. [Google Scholar] [CrossRef]
- Kashyap, S.J.; Sharma, P.K.; Garg, V.K.; Dudhe, R.; Kumar, N. Review on synthesis and various biological potential of thiazolopyrimidine derivatives. J. Adv. Sci. Res. 2011, 2, 18–24. [Google Scholar]
- Kajal, A.; Bala, S.; Sharma, N.; Kamboj, S.; Saini, V. Mannich Bases: An Important Pharmacophore in Present Scenario. Int. J. Med. Chem. 2014, 2014, 69–75. [Google Scholar]
- Gali, R.; Banothu, J.; Porika, M.; Velpula, R.; Hnamte, S.; Bavantula, R.; Busi, S. Indolylmethylene benzo[h]thiazolo [2,3-b]quinazolinones: Synthesis, characterization and evaluation of anticancer and antimicrobial activities. Bioorg. Med. Chem. 2014, 24, 4239–4242. [Google Scholar] [CrossRef]
- Studzińska, R.; Kołodziejska, R.; Redka, M.; Modzelewska-Banachiewicz, B.; Augustyńska, B. Lipophilicity study of thiazolo[3,2-a]pyrimidine derivatives as potential bioactive agents. J. Braz. Chem. Soc. 2016, 27, 1587–1593. [Google Scholar] [CrossRef]
- Zhou, B.; Li, X.; Li, Y.; Xu, Y.; Zhang, Z.; Zhou, M.; Wang, R. Discovery and Development of 2H-thiazolo[3,2-a]pyrimidine Derivatives as General Inhibitors of Bcl-2 Family Proteins. ChemMedChem 2011, 6, 904–921. [Google Scholar] [CrossRef]
- Feng, Y.; Ding, X.; Chen, T.; Chen, L.; Liu, F.; Jia, X.; Wang, H. Design, synthesis, and interaction study of quinazoline2(1H)-thione derivatives as novel potential Bcl-xL inhibitors. J. Med. Chem. 2010, 53, 3465–3479. [Google Scholar] [CrossRef]
- Kolb, S.; Mondésert, O.; Goddard, M.L.; Jullien, D.; Villoutreix, B.O.; Ducommun, B.; Braud, E. Development of Novel Thiazolopyrimidines as CDC25B Phosphatase Inhibitors. ChemMedChem. 2009, 4, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.H.; Jun, K.Y.; Lee, E.; Kim, S.; Kwon, Y.; Kim, K.; Na, Y. Ethyl 2-(benzylidene)-7-methyl-3-oxo-2,3-dihydro-5H-thiazolo[3,2-a] pyrimidine-6-carboxylate analogues as a new scaffold for protein kinase casein kinase 2 inhibitor. Bioorganic Med. Chem. 2014, 22, 4553–4565. [Google Scholar] [CrossRef] [PubMed]
- Geist, J.G.; Lauw, S.; Illarionova, V.; Illarionov, B.; Fischer, M.; Gräwert, T.; Rohdich, F.; Eisenreich, W.; Kaiser, J.; Groll, M.; et al. Thiazolopyrimidine inhibitors of 2-methylerythritol 2,4-cyclodiphosphate synthase (IspF) from Mycobacterium tuberculosis and Plasmodium falciparum. ChemMedChem. 2010, 5, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zeng, S. Stereoselective metabolic and pharmacokinetic analysis of the chiral active components from herbal medicines. Curr. Pharm. Anal. 2010, 6, 39–52. [Google Scholar] [CrossRef]
- Jozwiak, K.; Lough, W.J.; Wainer, I.W. Drug Stereochemistry: Analytical Methods and Pharmacology, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Sekhon, B.S. Exploiting the power of stereochemistry in drugs: An overview of racemic and enantiopure drugs. J. Mod. Med. Chem. 2013, 1, 10–36. [Google Scholar] [CrossRef] [Green Version]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef]
- Solano, D.M.; Muñoz Solano, D.; Hoyos, P.; Hernáiz, M.J.; Alcántara, A.R.; Sánchez-Montero, J.M. Industrial biotransformations in the synthesis of building blocks leading to enantiopure drugs. Bioresour. Technol. 2012, 115, 196–207. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, D.R.; Wang-Iverson, D.B.; Tymiak, A.A. Enantioselective chromatography in drug discovery. Drug Discov. Today 2005, 10, 571–577. [Google Scholar] [CrossRef]
- Liu, X.G.; Feng, Y.Q.; Li, X.F.; Liang, Z.P. Ethyl 2-(2, 4-dichlorobenzylidene)-7-methyl-3-oxo-5-phenyl-2, 3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate dichloromethane solvate. Acta Crystallogr. Sect. E Struct. Rep. Online 2004, 60, 344–345. [Google Scholar] [CrossRef] [Green Version]
- Jotani, M.M.; Baldaniya, B.B. (2Z)-Ethyl 2-(4-chlorobenzylidene)-7-methyl-3-oxo-5-phenyl-2, 3-dihydro-5H-1, 3thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2006, 62, 5871–5873. [Google Scholar] [CrossRef]
- Jotani, M.M.; Baldaniya, B.B.; Jasinski, J.P. Crystal Structure of Ethyl (2Z, 5R)-2-benzylidene-7-methyl-3-oxo-5-phenyl-2, 3-dihydro-5H-[1,3] Thiazolo[3,2-a]Pyrimidine-6-carboxylate. J. Chem. Crystallogr. 2009, 39, 898–901. [Google Scholar] [CrossRef]
- Jotani, M.M.; Baldaniya, B.B. Ethyl 2-[(Z)-3-chlorobenzylidene]-7-methyl-3-oxo-5-phenyl-2, 3-dihydro-5H-1, 3-thiazolo[3,2-a] pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64, 739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.G.; Feng, Y.Q.; Li, X.F.; Gao, B. Ethyl 5-(2, 6-dichlorophenyl)-7-methyl-2-(1-naphthylmethylene)-3-oxo-2, 3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2004, 60, 464–465. [Google Scholar] [CrossRef]
- Banu, N.A.; Raju, V.B. Ethyl 2-(4-carboxybenzylidene)-7-methyl-3-oxo-5-phenyl-2, 3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate–N, N-dimethylformamide. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Z.H.; Zhou, N.B.; He, B.H.; Li, X.F. Ethyl 5-(4-chlorophenyl)-2-[(Z)-(methoxycarbonyl) methylene]-7-methyl-3-oxo-3, 5-dihydro-2H-thiazolo[3, 2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, 375. [Google Scholar] [CrossRef]
- Jotani, M.M.; Baldaniya, B.B.; Tiekink, E.R. Ethyl 2-(2-acetoxybenzylidene)-7-methyl-3-oxo-5-phenyl-2, 3-dihydro-5H-1, 3-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, 762–763. [Google Scholar] [CrossRef]
- Baldaniya, B.B.; Jotani, M.M. Crystal Structure of Ethyl (2Z)-2-(4-acetyloxybenzylidene)-7-methyl-3-oxo-5-phenyl-2, 3-dihydro-5H-[1,3] thiazolo[3,2-a]pyrimidine-6-carboxylate. Anal. Sci. X-ray Struct. Anal. Online 2008, 24, 217–218. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.H. (2Z)-Ethyl 5-(4-methoxyphenyl)-7-methyl-3-oxo-2-(3, 4, 5-trimethoxybenzylidene)-3, 5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, 235. [Google Scholar] [CrossRef]
- Krishnamurthy, M.S.; Nagarajaiah, H.; Begum, N.S. Crystal structure of ethyl 5-(3-fluorophenyl)-2-[(4-fluorophenyl) methylidene]-7-methyl-3-oxo-2H, 3H, 5H-[1,3] thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2014, 70, 1187–1188. [Google Scholar] [CrossRef] [Green Version]
- Jotani, M.M.; Baldaniya, B.B.; Jasinski, J.P. Ethyl (2Z)-2-(3-methoxybenzylidene)-7-methyl-3-oxo-5-phenyl-2, 3-dihydro-5H-1, 3-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, 599–600. [Google Scholar] [CrossRef] [Green Version]
- Nagarajaiah, H.; Begum, N.S. Structural modifications leading to changes in supramolecular aggregation of thiazolo[3,2-a]pyrimidines: Insights into their conformational features. J. Chem. Sci. 2014, 126, 1347–1356. [Google Scholar] [CrossRef]
- Chen, X.Y.; Wang, H.C.; Zhang, Q.; Song, Z.J.; Zheng, F.Y. (Z)-Ethyl 2-(2, 4-dimethylbenzylidene)-7-methyl-3-oxo-5-phenyl-3, 5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, 127. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wu, X.X.; Shen, X.Q.; Tang, L.G.; Li, X.K. Ethyl 7-methyl-2-((1-methyl-1H-pyrrol-2-yl) methylene)-3-oxo-5-phenyl-3, 5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, 3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Yathirajan, H.S.; Mithun, A.; Bindya, S.; Narayana, B. Ethyl 7-methyl-2-[4-(methylsulfanyl) benzylidene]-5-[4-(methylsulfanyl) phenyl]-3-oxo-2, 3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2007, 63, 1224–1225. [Google Scholar] [CrossRef]
- Jotani, M.M.; Baldaniya, B.B. Ethyl (2Z)-2-(2-chlorobenzylidene)-7-methyl-3-oxo-5-phenyl-2, 3-dihydro-5H-1, 3-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2007, 63, 1937–1939. [Google Scholar] [CrossRef]
- Nagarajaiah, H.; Khazi, I.A.M.; Begum, N.S. Synthesis of some new derivatives of thiazolopyrimidines and hydrolysis of its arylidene derivative. J. Chem. Sci. 2015, 127, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.G.; Hu, J.; Zhang, Y.L.; Zhang, J.; Yang, S.L. Ethyl (Z)-2-(2-fluorobenzylidene)-7-methyl-3-oxo-5-phenyl-3, 5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2011, 67, 3009. [Google Scholar] [CrossRef]
- Banu, N.A.; Bheema Raju, V. Ethyl 7-methyl-3-oxo-5-phenyl-2-(2, 4, 6-trimethoxybenzylidene)-2, 3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, 1213. [Google Scholar] [CrossRef]
- Krishnamurthy, M.S.; Begum, N.S. Crystal structure of ethyl 2-(2-fluorobenzylidene)-5-(4-fluorophenyl)-7-methyl-3-oxo-2, 3-dihydro-5H-1, 3-thiazolo[3,2-a]pyrimidine-6-carboxylate. Acta Crystallogr. Sect. E Struct. Rep. Online 2014, 70, 1270–1271. [Google Scholar] [CrossRef] [Green Version]
- Bredikhin, A.A.; Bredikhina, Z.A.; Gubaidullin, A.T.; Litvinov, I.A. Crystallization of chiral compounds. 2. Propranolol: Free base and hydrochloride. Russ. Chem. Bull. 2003, 52, 853–861. [Google Scholar] [CrossRef]
- Lashmanova, E.A.; Rybakov, V.B.; Shiryaev, A.K. Synthesis of adamantylated pyrimidines using the Biginelli reaction. Synthesis 2016, 48, 3965–3970. [Google Scholar]
- Shiryaev, A.K.; Baranovskaya, N.S.; Eremin, M.S. Synthesis of 5H-thiazolo[3,2-a]pyrimidines. Chem. Heterocycl. Compd. 2012, 48, 1662–1667. [Google Scholar] [CrossRef]
- Shiryaev, A.K.; Kolesnikova, N.G.; Kuznetsova, N.M.; Lashmanova, E.A. Alkylation of tetrahydropyrimidine-2-thions with ethyl chloroacetate. Chem. Heterocycl. Compd. 2013, 49, 1812–1817. [Google Scholar] [CrossRef]
- Lashmanova, E.A.; Kirdyashkina, A.I.; Slepukhin, P.A.; Shiryaev, A.K. Oxidation of thiazolo[3,2-a]pyrimidin-3 (2H)-ones with DMSO and Lawesson’s reagent. Tetrahedron Lett. 2018, 59, 1099–1103. [Google Scholar] [CrossRef]
- Lashmanova, E.A.; Agarkov, A.S.; Rybakov, V.B.; Shiryaev, A.K. Rearrangement of thiazolo[3,2-a]pyrimidines into triazolo [4,3-a]pyrimidines induced by C=N bond reduction. Chem. Heterocycl. Compd. 2019, 55, 1217–1221. [Google Scholar] [CrossRef]
- Lazarenko, V.A.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Burlov, A.S.; Koshchienko, Y.V.; Vlasenko, V.G.; Khrustalev, V.N. High-throughput small-molecule crystallography at the ‘Belok’beamline of the Kurchatov synchrotron radiation source: Transition metal complexes with azomethine ligands as a case study. Crystals 2017, 7, 325. [Google Scholar] [CrossRef] [Green Version]
- Svetogorov, R.D.; Dorovatovskii, P.V.; Lazarenko, V.A. Belok/XSA diffraction beamline for studying crystalline samples at Kurchatov Synchrotron Radiation Source. Cryst. Res. Technol. 2020, 55, 1900184. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Streek, J.V.D. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Flack, H.D.; Bernardinelli, G. Reporting and evaluating absolute-structure and absolute-configuration dete rminations. J. Appl. Crystallogr. 2000, 33, 1143–1148. [Google Scholar] [CrossRef] [Green Version]
- Collet, A.; Brienne, M.J.; Jacques, J. Optical resolution by direct crystallization of enantiomer mixtures. Chem. Rev. 1980, 80, 215–230. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Sasaki, T.; Ida, Y.; Hisaki, I.; Tsuzuki, S.; Tohnai, N.; Coquerel, G.; Sato, H.; Miyata, M. Construction of Chiral Polar Crystals from Achiral Molecules by Stacking Control of Hydrogen-Bonded Layers Using Type II Halogen Bonds. Cryst. Growth Des. 2016, 16, 1626–1635. [Google Scholar] [CrossRef]










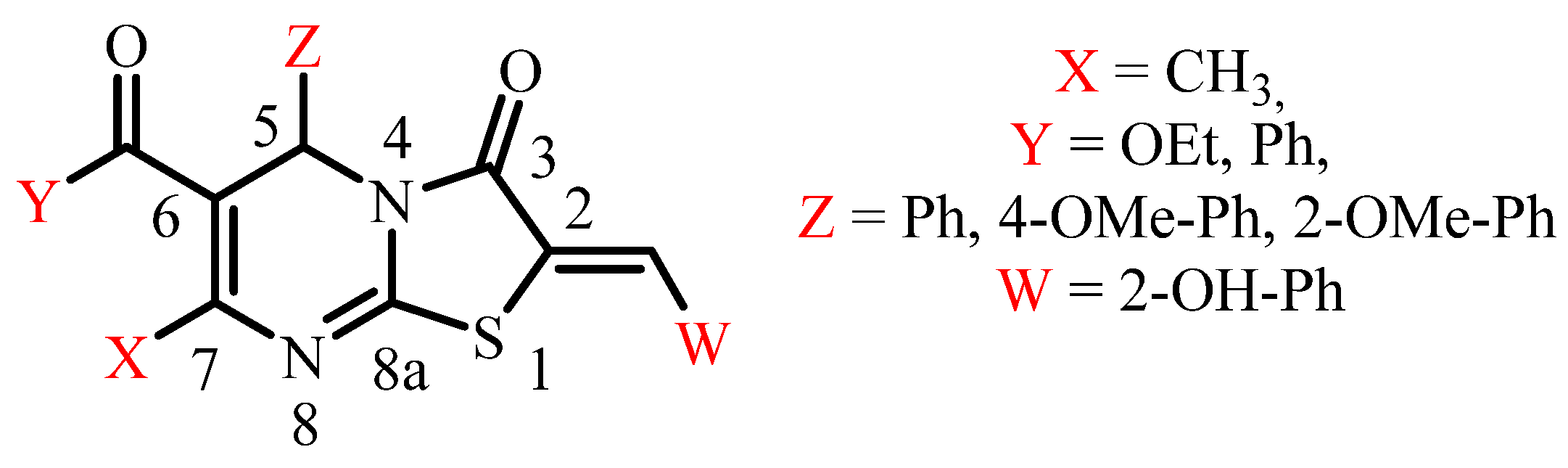{kind=link}
{kind=link}
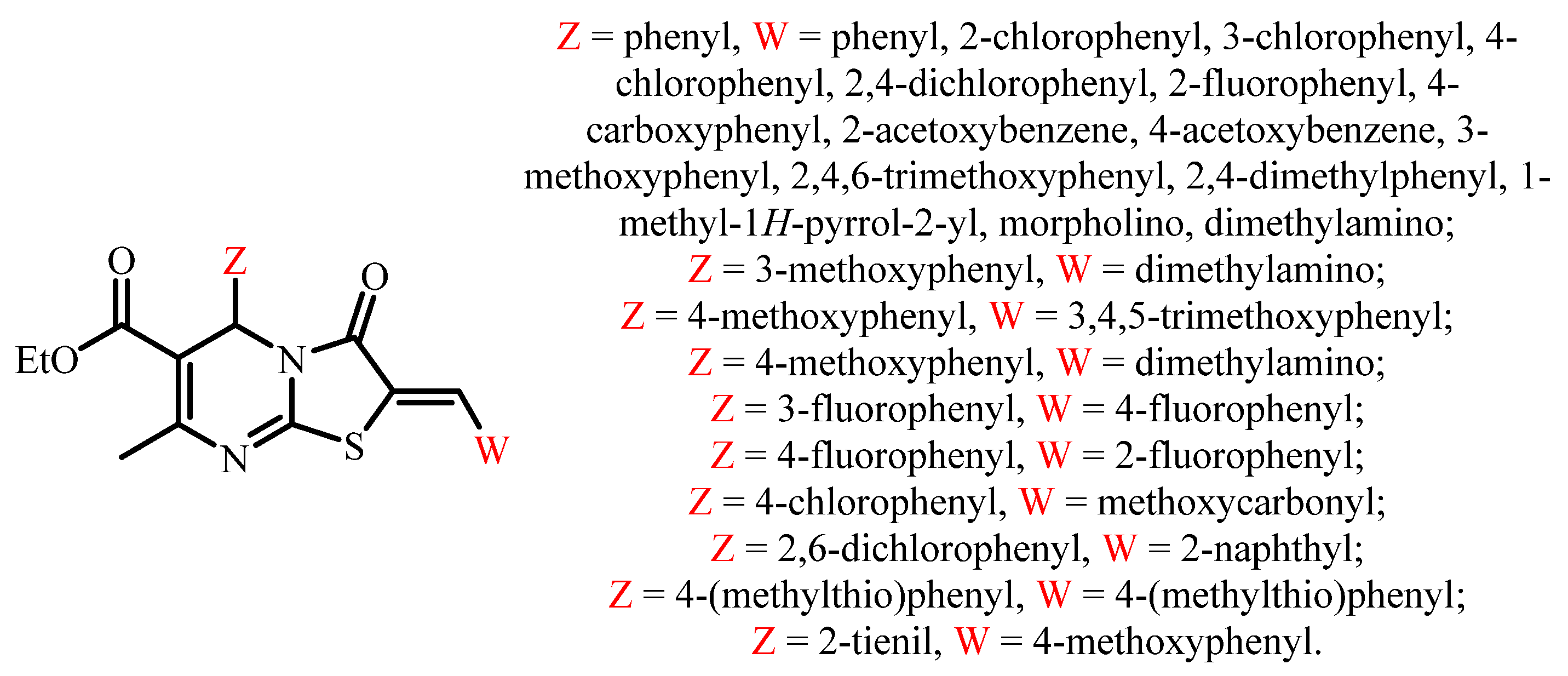{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 (from Ethanol) | 2a (from Ethanol) | 2b (from DMSO) | 3 (from Ethanol) |
|---|---|---|---|---|
| Molecular formula | C23H20N2O4 S, C2H6O | C24H22N2O5S | C24H22N2O5S, C2H6OS | C24H22N2O5S |
| Formula | C25H26N2O5S | C24H22N2O5S | C26H28N2O6S2 | C24H22N2O5S |
| Formula Weight | 466.54 | 450.50 | 528.62 | 450.50 |
| Crystal System | monoclinic | triclinic | monoclinic | orthorhombic |
| Space group | P21 | P-1 (P1bar) | P21/c | P212121 |
| Cell parameters | a = 11.7398(16) Å, b = 21.619(3) Å, c = 9.6209(13) Å; β = 108.231(4)° | a = 6.6700(13) Å, b = 11.210(2) Å, c = 14.630(3) Å; α = 77.42(3)° β = 77.45(3)° γ = 78.96(3)° | a = 9.6076(8) Å, b = 17.8987(15) Å, c = 14.7477(12) Å; β = 101.158(3)° | a = 7.7520(16) Å, b = 12.147(2) Å, c = 22.309(5) Å; |
| V [Å3] | 2319.2(6) Å3 | 1030.1(4) Å3 | 2488.1(4) Å3 | 2100.7(7) Å3 |
| Z and Z′ | 4 and 2 | 2 and 1 | 4 and 1 | 4 and 1 |
| D(calc) [g/cm3] | 1.336 | 1.452 | 1.411 | 1.424 Γ·CM−3 |
| λ (Å) | (MoKα) 0.71073 | 0.7450 | (MoKα) 0.71073 | 0.7450 |
| μ [/mm] | 0.179 MM−1 | 0.221 MM−1 | 0.260 MM−1 | 0.217 MM−1 |
| F(000) | 984 | 472 | 1112 | 944 |
| Theta Min-Max [Deg] | 1.9–26.5° | 2.0–30.0° | 2.2–30.0° | 1.9–26.3° |
| Reflections measured | 75,170 | 19,400 | 118,981 | 11,919 |
| Independent reflections | 9502 | 5168 | 7251 | 3699 |
| Observed reflections [I > 2σ(I)] | 8409 | 4893 | 6494 | 1821 |
| Goodness of fit | 0.997 | 1.03 | 1.04 | 0.986 |
| R [I > 2σ(I)] | R1 = 0.0277, wR2 = 0.0702 | R1 = 0.0353, wR2 = 0.0936 | R1 = 0.0307, wR2 = 0.0818 | R1 = 0.0848, wR2 = 0.1582 |
| R (all reflections) | R1 = 0.0341, wR2 = 0.0719 | R1 = 0.0368, wR2 = 0.0951 | R1 = 0.0352, wR2 = 0.845 | R1 = 0.1879, wR2 = 0.2041 |
| Max. and Min. Resd. Dens. [e/Å−3] | 0.216 and −0.182 e Å−3 | 0.41 and −0.24 e Å−3 | 0.52 и −0.34 e Å−3 | 0.32 and −0.34 e Å−3 |
| Flack parameter | 0.48(5) (racemic twin, BASF 0.4785) | - | - | 0.4(3) |
| Depositor numbers in CCDC | 2,155,259 | 2,155,261 | 2,155,263 | 2,155,264 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarkov, A.S.; Litvinov, I.A.; Gabitova, E.R.; Ovsyannikov, A.S.; Dorovatovskii, P.V.; Shiryaev, A.K.; Solovieva, S.E.; Antipin, I.S. Crystalline State Hydrogen Bonding of 2-(2-Hydroxybenzylidene)Thiazolo[3,2-a]Pyrimidines: A Way to Non-Centrosymmetric Crystals. Crystals 2022, 12, 494. https://doi.org/10.3390/cryst12040494
Agarkov AS, Litvinov IA, Gabitova ER, Ovsyannikov AS, Dorovatovskii PV, Shiryaev AK, Solovieva SE, Antipin IS. Crystalline State Hydrogen Bonding of 2-(2-Hydroxybenzylidene)Thiazolo[3,2-a]Pyrimidines: A Way to Non-Centrosymmetric Crystals. Crystals. 2022; 12(4):494. https://doi.org/10.3390/cryst12040494
Chicago/Turabian StyleAgarkov, Artem S., Igor A. Litvinov, Elina R. Gabitova, Alexander S. Ovsyannikov, Pavel V. Dorovatovskii, Andrey K. Shiryaev, Svetlana E. Solovieva, and Igor S. Antipin. 2022. "Crystalline State Hydrogen Bonding of 2-(2-Hydroxybenzylidene)Thiazolo[3,2-a]Pyrimidines: A Way to Non-Centrosymmetric Crystals" Crystals 12, no. 4: 494. https://doi.org/10.3390/cryst12040494
APA StyleAgarkov, A. S., Litvinov, I. A., Gabitova, E. R., Ovsyannikov, A. S., Dorovatovskii, P. V., Shiryaev, A. K., Solovieva, S. E., & Antipin, I. S. (2022). Crystalline State Hydrogen Bonding of 2-(2-Hydroxybenzylidene)Thiazolo[3,2-a]Pyrimidines: A Way to Non-Centrosymmetric Crystals. Crystals, 12(4), 494. https://doi.org/10.3390/cryst12040494