
Periodicity of Superatomic Hybrid Orbitals in Substituted Superatoms and Superatomic-like X@Ga12 (X = Li~Kr) Clusters


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
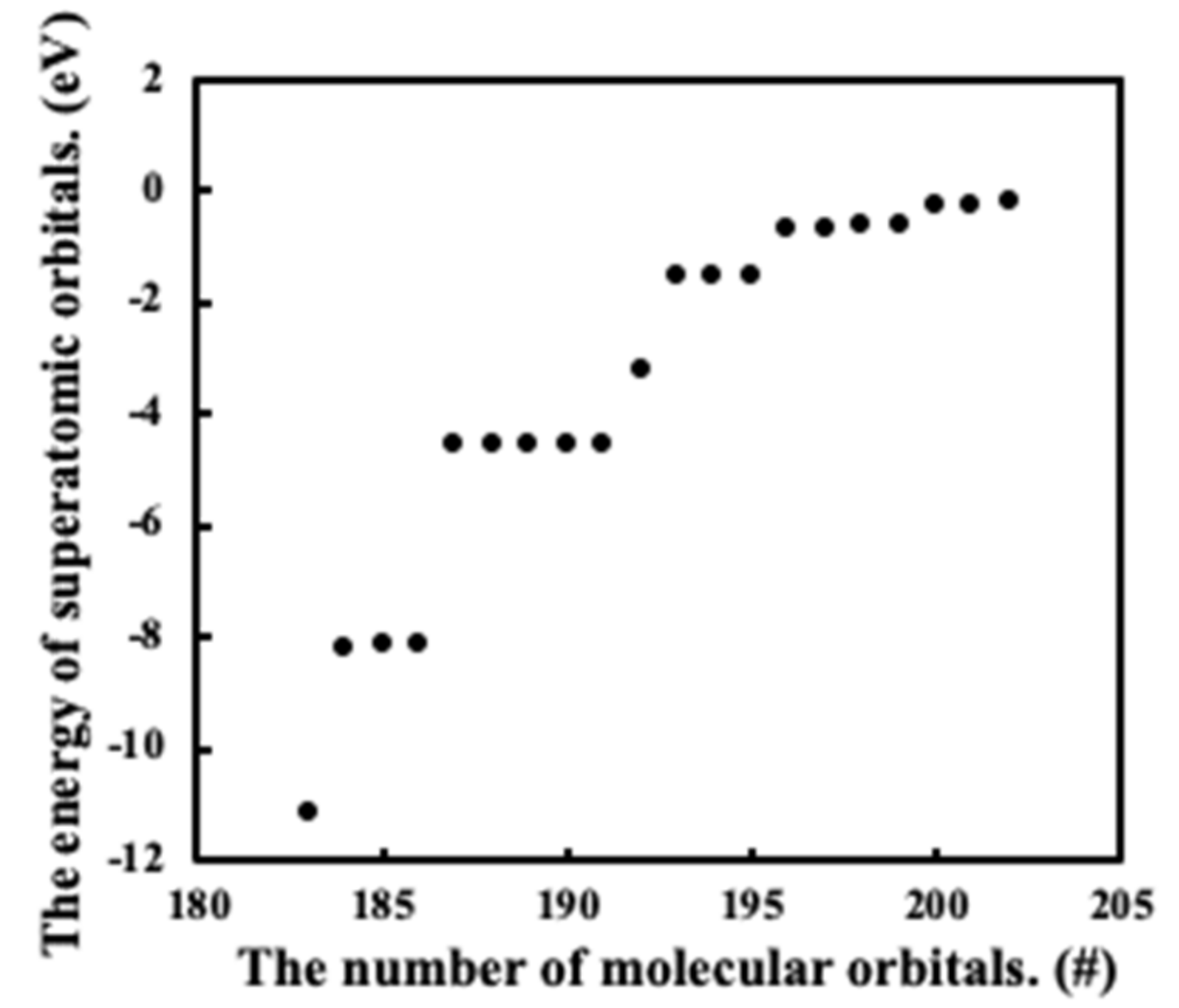
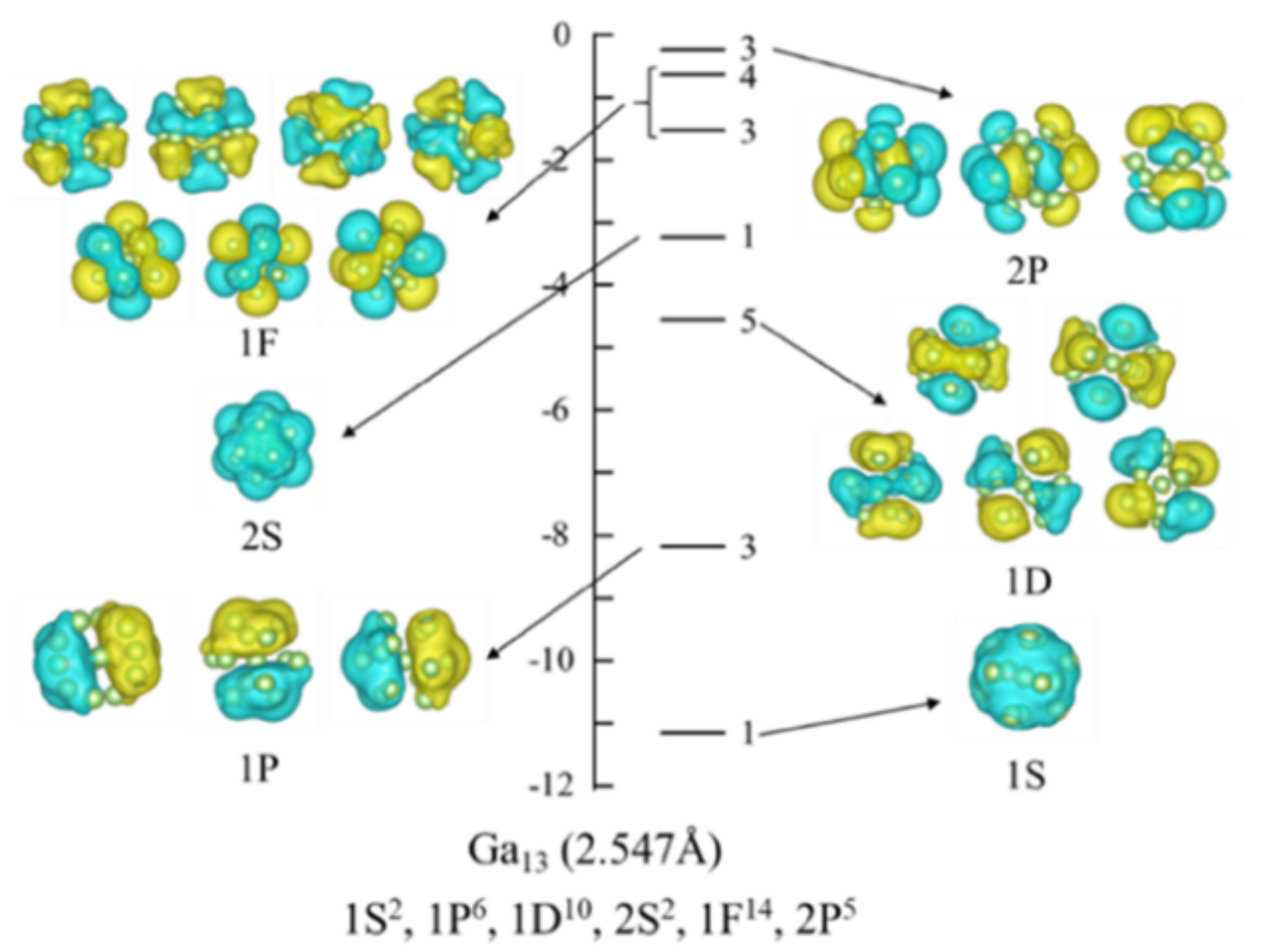
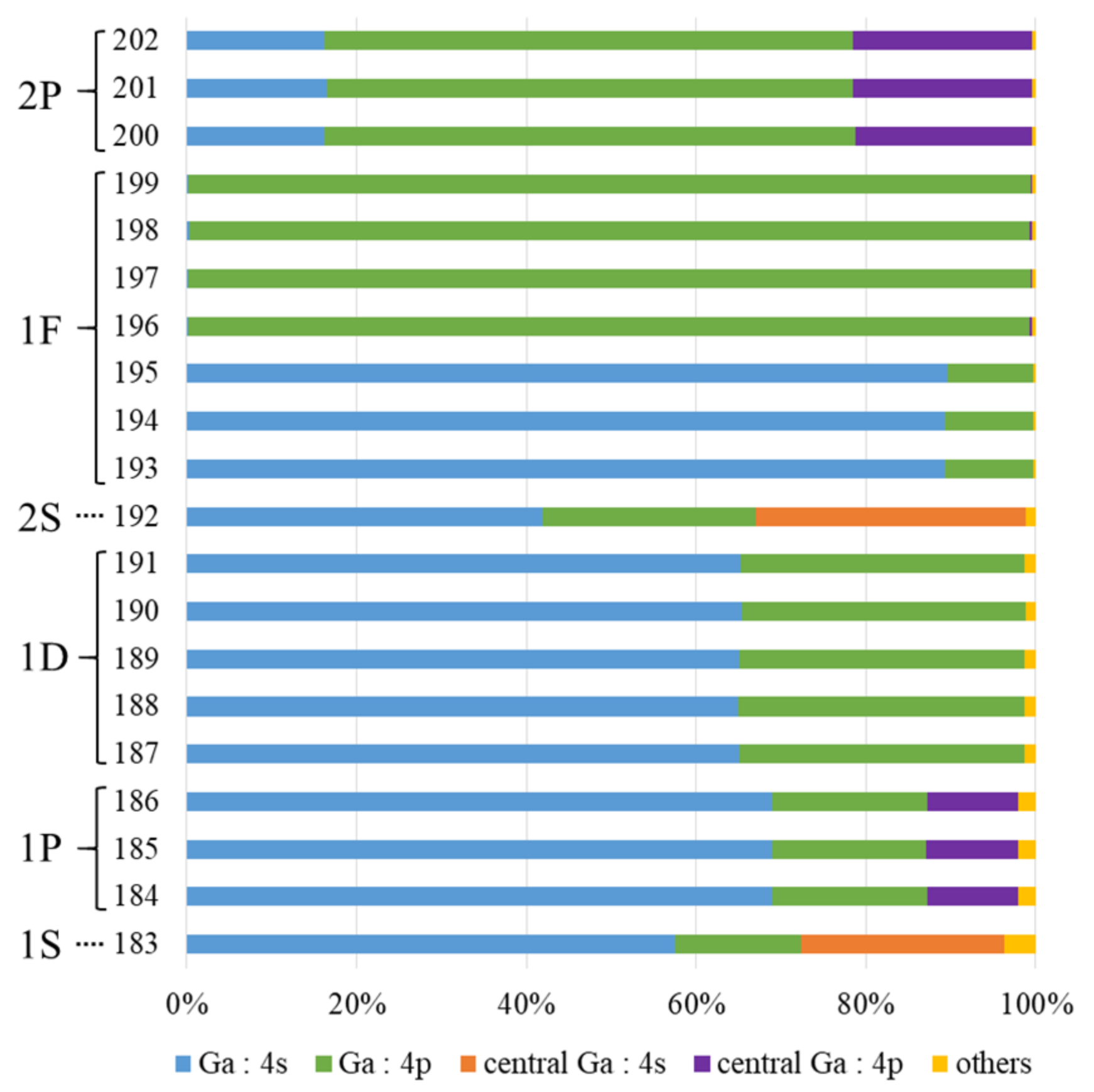
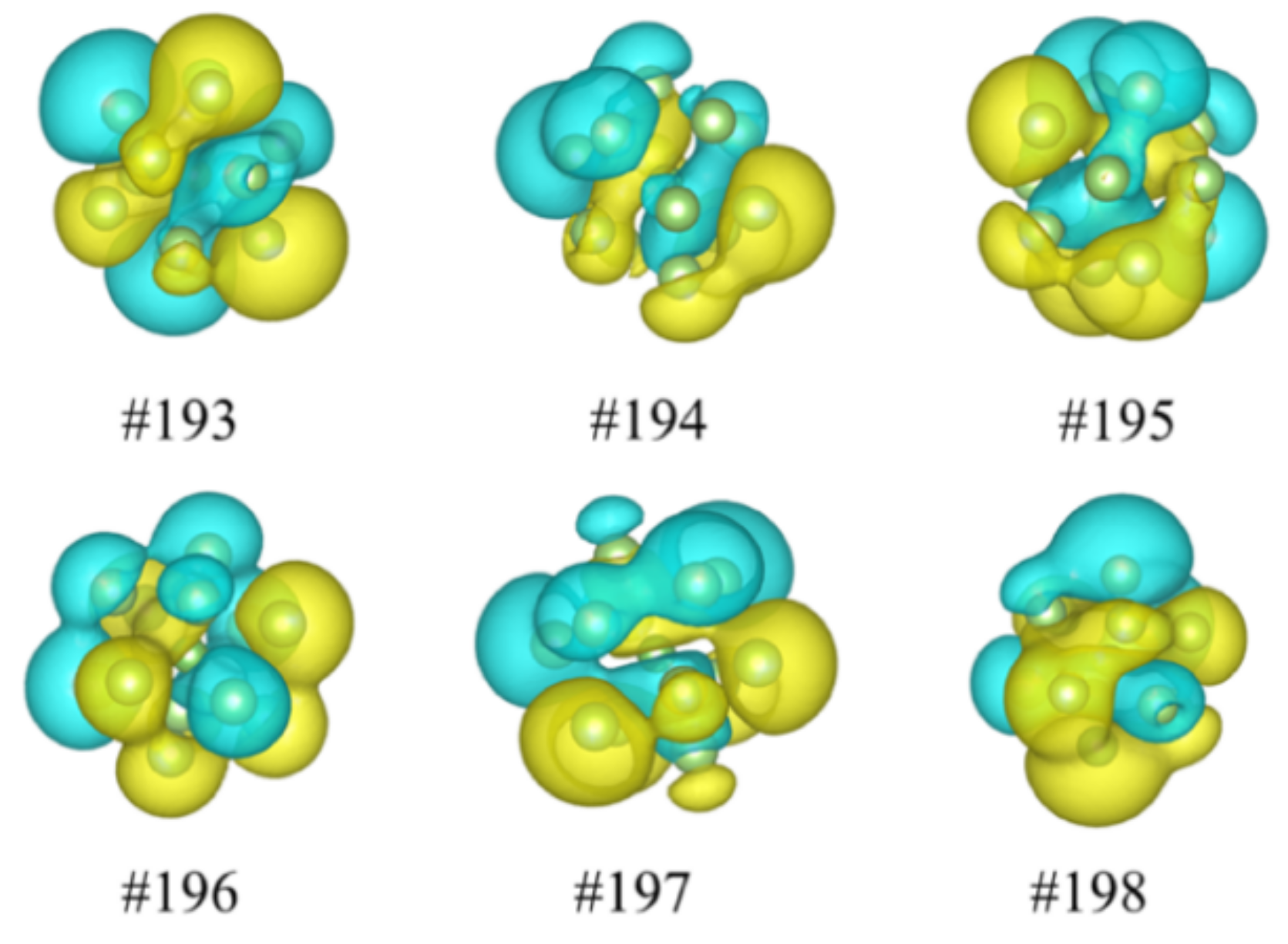
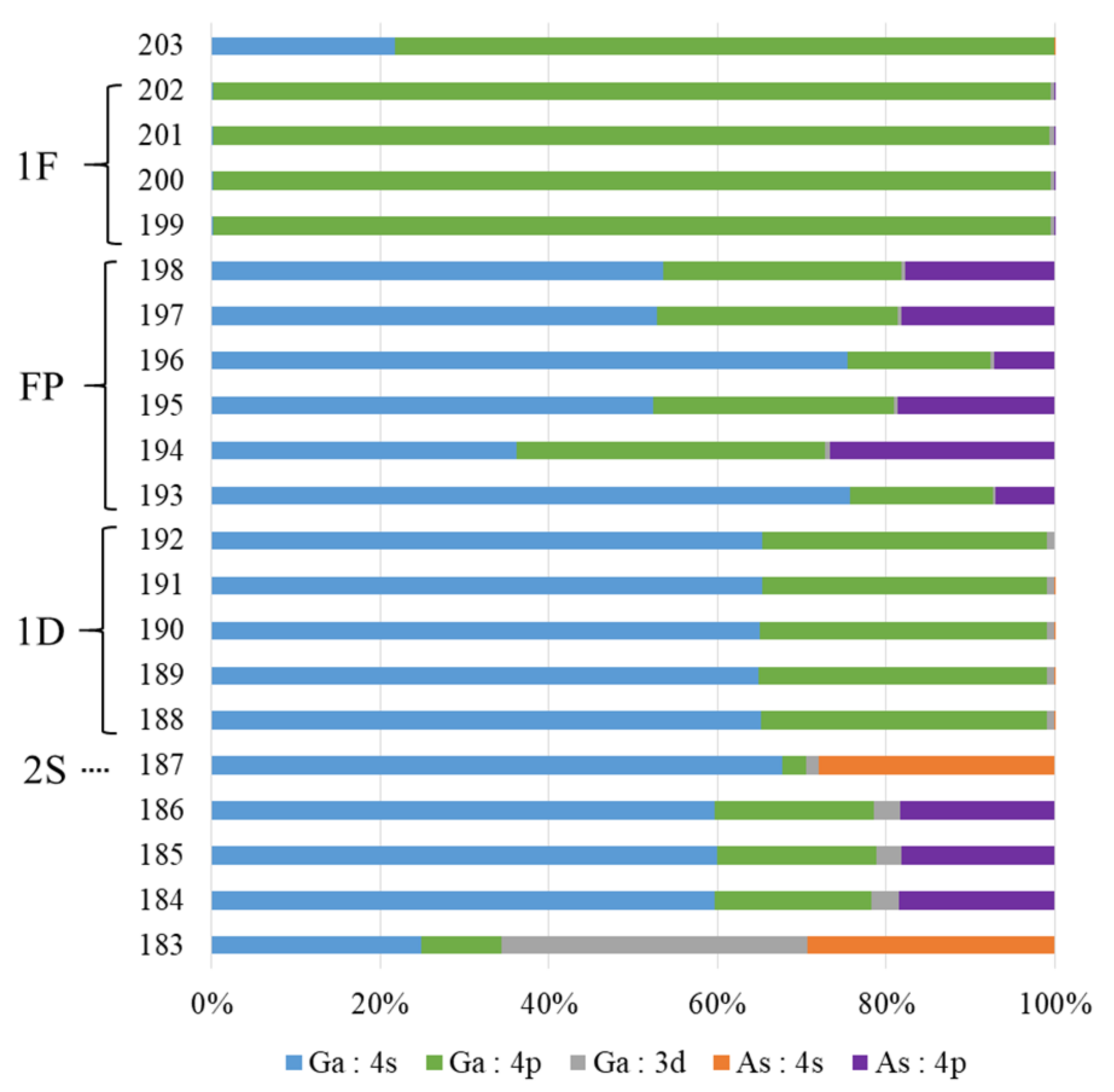
3.1. Wave Functions of Superatomic Orbitals and Hybridization Ratio of Atomic Orbitals in the Superatom Ga13
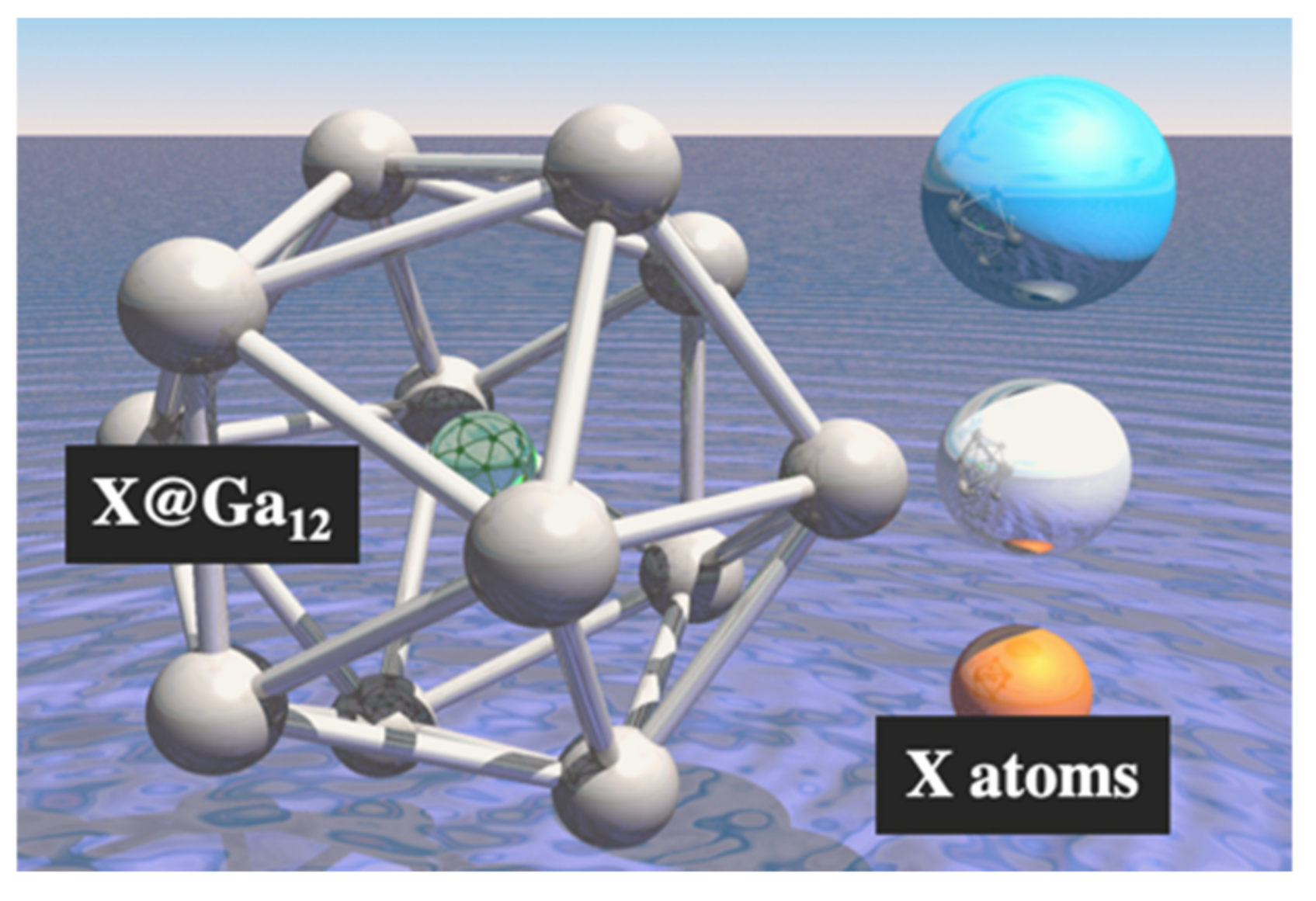
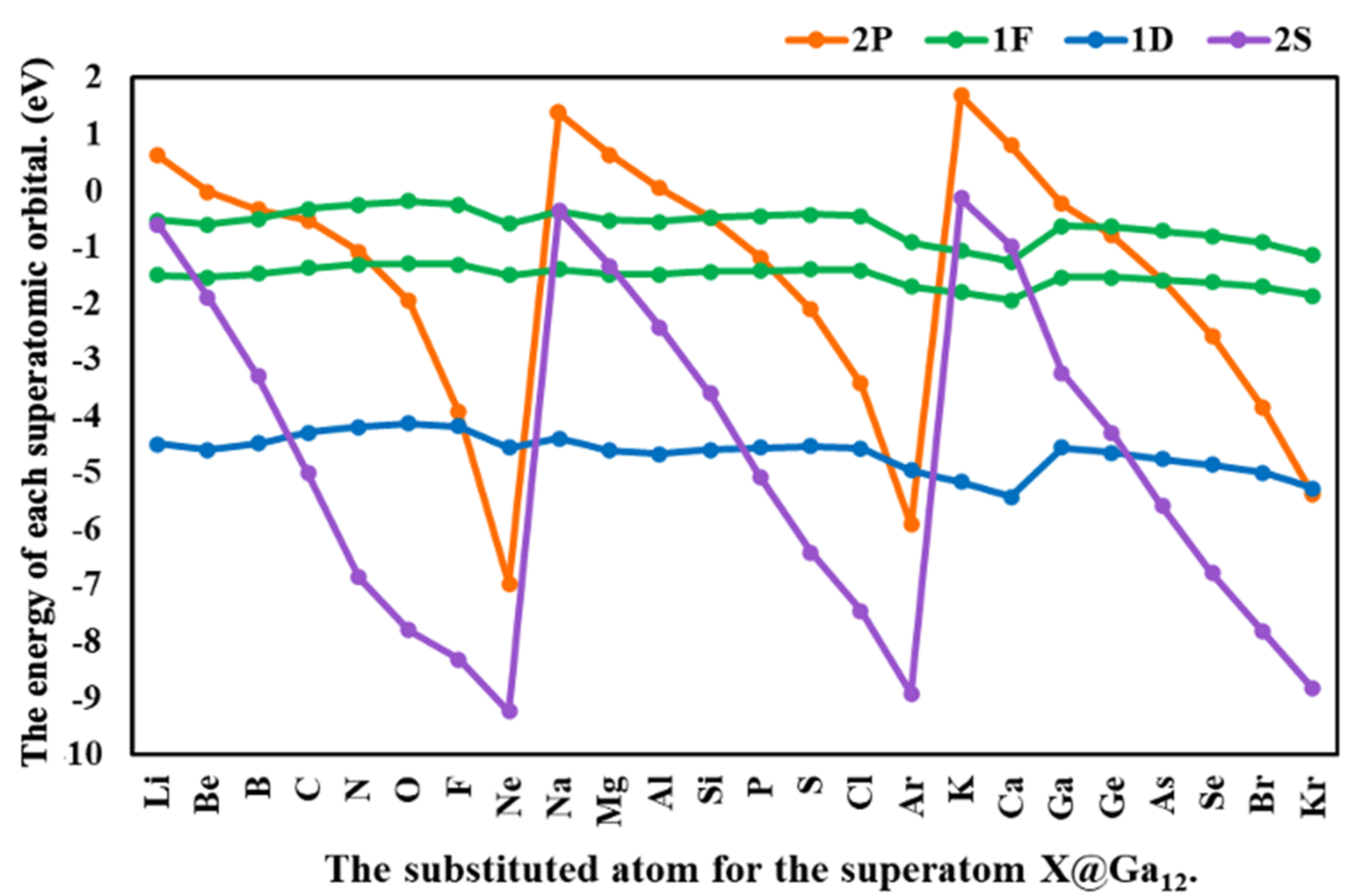
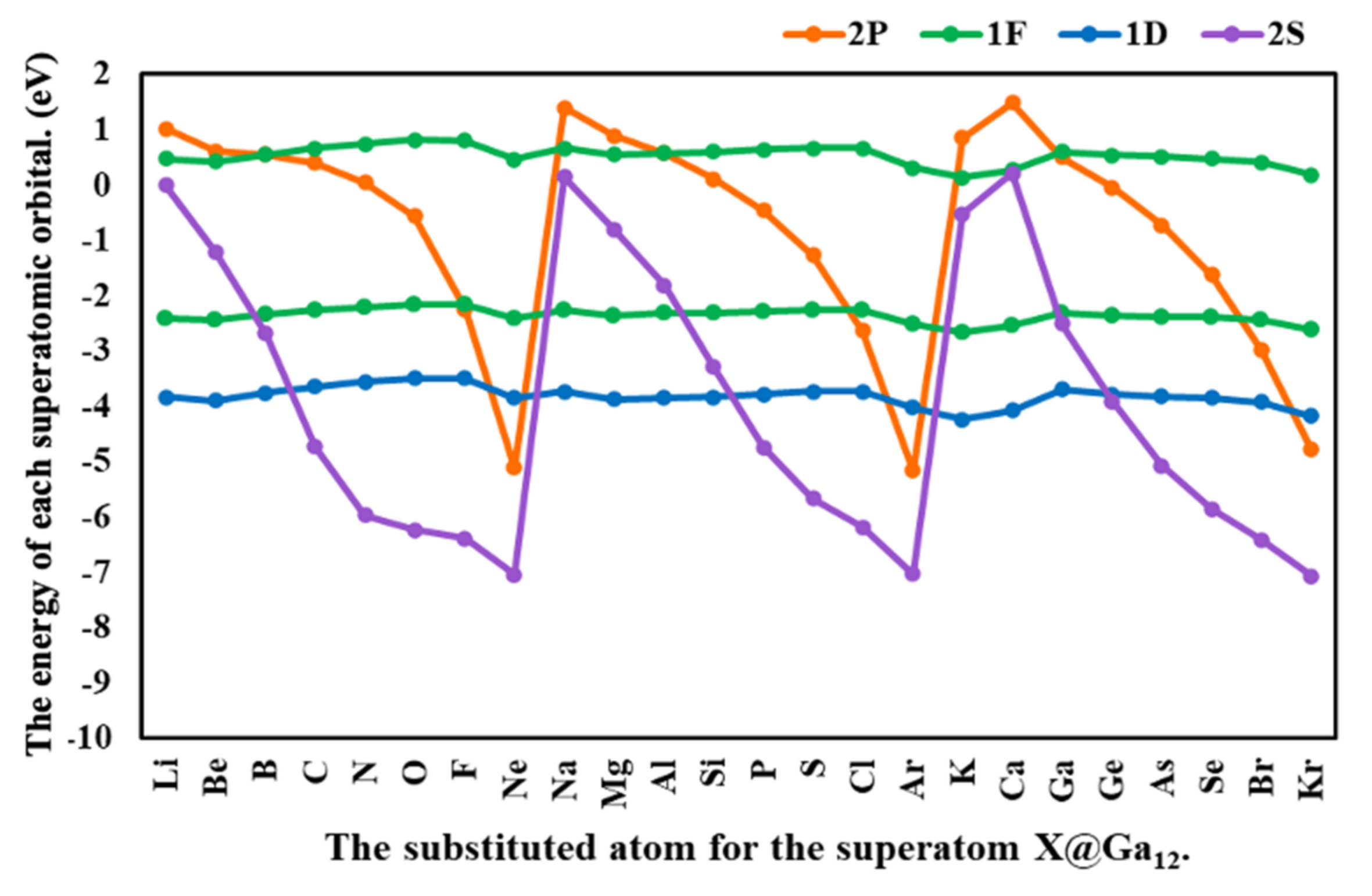
3.2. The Effects of the Atom Substituted for the Central Atom in the Ga13 Cluster (X@Ga12)
3.3. The Limit of the Conditions for Applying the Jellium Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Heer, W.A. The physics of simple metal clusters: Experimental aspects and simple models. Rev. Mod. Phys. 1993, 65, 611. [Google Scholar] [CrossRef]
- Leuchtner, R.E.; Harms, A.C.; Castleman, A.W., Jr. Thermal metal cluster anion reactions: Behavior of aluminum clusters with oxygen. J. Chem. Phys. 1989, 91, 2753–2754. [Google Scholar] [CrossRef]
- Luo, Z.; Grover, C.J.; Reber, A.C.; Khanna, S.N.; Castleman, A.W., Jr. Probing the magic numbers of aluminum–magnesium cluster anions and their reactivity toward oxygen. J. Am. Chem. Soc. 2013, 135, 4307–4313. [Google Scholar] [CrossRef] [PubMed]
- Brack, M. The physics of simple metal clusters: Self-consistent Jellium model and semiclassical approaches. Rev. Mod. Phys. 1993, 65, 677. [Google Scholar] [CrossRef] [Green Version]
- Knight, W.D.; Clemenger, K.; de Heer, W.A.; Saunders, W.A.; Chou, M.Y.; Cohen, M.L. Electronic shell structure and abundances of sodium clusters. Phys. Rev. Lett. 1984, 52, 2141. [Google Scholar] [CrossRef]
- Luo, Z.; Castleman, A.W. Special and general superatoms. Acc. Chem. Res. 2014, 47, 2931–2940. [Google Scholar] [CrossRef]
- Bergeron, D.E.; Roach, P.J.; Castleman, A.W.; Jones, N.O.; Khanna, S.N. Al cluster superatoms as halogens in polyhalides and as alkaline earths in iodide salts. Science 2005, 307, 231–235. [Google Scholar] [CrossRef]
- Bergeron, D.E.; Castleman, A.W.; Morisato, T.; Khanna, S.N. Formation of Al13I-: Evidence for the superhalogen character of Al13. Science 2004, 304, 84–87. [Google Scholar] [CrossRef]
- Bergeron, D.E.; Castleman, A.W., Jr.; Morisato, T.; Khanna, S.N. Formation and properties of halogenated aluminum clusters. J. Chem. Phys. 2004, 121, 10456–10466. [Google Scholar] [CrossRef]
- Schnöckel, H.; Köhnlein, H. Synthesis and structure of metalloid aluminum clusters—Intermediates on the way to the elements. Polyhedron 2002, 21, 489–501. [Google Scholar] [CrossRef]
- Luo, Z.; Berkdemir, C.; Smith, J.C.; Castleman, A.W., Jr. Cluster reaction of [Ag8]−/[Cu8]− with chlorine: Evidence for the harpoon mechanism? Chem. Phys. Lett. 2013, 582, 24–30. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Wang, H.; Lim, A.; Gantefoer, G.; Bowen, K.H.; Reveles, J.U.; Khanna, S.N. On the existence of designer magnetic superatoms. Acc. Chem. Res. 2013, 135, 4856–4861. [Google Scholar] [CrossRef] [PubMed]
- Reber, A.C.; Khanna, S.N. Superatoms: Electronic and geometric effects on reactivity. Acc. Chem. Res. 2017, 50, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.I. Theoretical practice: The Bohm-Pines quartet. Perspect. Sci. 2006, 14, 457–524. [Google Scholar] [CrossRef]
- Khanna, S.N.; Jena, P. Atomic clusters: Building blocks for a class of solids. Phys. Rev. B 1995, 51, 13705. [Google Scholar] [CrossRef]
- Castleman, A.W., Jr.; Khanna, S.N. Clusters, superatoms, and building blocks of new materials. J. Phys. Chem. C 2009, 113, 2664–2675. [Google Scholar] [CrossRef]
- Kambe, T.; Haruta, N.; Imaoka, T.; Yamamoto, K. Solution-phase synthesis of Al13− using a dendrimer template. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Narouz, M.R.; Takano, S.; Lummis, P.A.; Levchenko, T.I.; Nazemi, A.; Kaappa, S.; Malola, S.; Yousefalizadeh, G.; Calhoun, L.A.; Stamplecoskie, K.G.; et al. Robust, highly luminescent Au13 superatoms protected by N-heterocyclic carbenes. J. Am. Chem. Soc. 2019, 141, 14997–15002. [Google Scholar] [CrossRef]
- Zhu, M.; Aikens, C.M.; Hendrich, M.P.; Gupta, R.; Qian, H.; Schatz, G.C.; Jin, R. Reversible switching of magnetism in thiolate-protected Au25 superatoms. J. Am. Chem. Soc. 2009, 131, 2490–2492. [Google Scholar] [CrossRef]
- Negishi, Y.; Kurashige, W.; Kobayashi, Y.; Yamazoe, S.; Kojima, N.; Seto, M.; Tsukuda, T. Formation of a Pd@ Au12 superatomic core in Au24Pd1(SC12H25)18 probed by 197Au Mossbauer and Pd K-Edge EXAFS spectroscopy. J. Phys. Chem. Lett. 2013, 4, 3579–3583. [Google Scholar] [CrossRef]
- Chen, J.; Yang, H.; Wang, J.; Cheng, S.B. Revealing the effect of the oriented external electronic field on the superatom-polymeric Zr3O3 cluster: Superhalogen modulation and spectroscopic characteristics. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 237, 118400. [Google Scholar] [CrossRef]
- Reber, A.C.; Khanna, S.N.; Castleman, A.W. Superatom compounds, clusters, and assemblies: Ultra alkali motifs and architectures. J. Am. Chem. Soc. 2007, 129, 10189–10194. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Giri, S. Stability, reactivity, and aromaticity of compounds of a multivalent superatom. J. Phys. Chem. A 2007, 111, 11116–11121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jena, P.; Khanna, S.N.; Rao, B.K. Designing clusters as superelements. Surf. Rev. Lett. 1996, 3, 993–999. [Google Scholar] [CrossRef]
- Jena, P. Beyond the periodic table of elements: The role of superatoms. J. Phys. Chem. Lett. 2013, 4, 1432–1442. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M. Open-shell jellium aromaticity in metal clusters. Chem. Commun. 2019, 55, 5559–5562. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Slater, J.C. A simplification of the Hartree-Fock method. Phys. Rev. 1951, 81, 385. [Google Scholar] [CrossRef]
- Adachi, H.; Tsukuda, M.; Satoko, C. Discrete variational Xα cluster calculations. I. Application to metal clusters. J. Phys. Soc. Jpn. 1978, 45, 875–883. [Google Scholar] [CrossRef]
- Satoko, C.; Tsukada, M.; Adachi, H. Discrete variational Xα cluster calculations. II. Application to the surface electronic structure of MgO. J. Phys. Soc. Jpn. 1978, 45, 1333–1340. [Google Scholar] [CrossRef]
- Adachi, H.; Shiokawa, S.; Tsukada, M.; Satoko, C.; Sugano, S. Discrete variational X α cluster calculations. III. Application to transition metal complexes. J. Phys. Soc. Jpn. 1979, 47, 1528–1537. [Google Scholar] [CrossRef]
- Adachi, H.; Taniguchi, K. Discrete variational Xα cluster calculations. IV. Application to X-ray emission study. J. Phys. Soc. Jpn. 1980, 49, 1944–1953. [Google Scholar] [CrossRef]
- Adachi, H.; Mukoyama, T.; Kawai, J. (Eds.) Hartree-Fock-Slater Method for Materials Science: The DV-X Alpha Method for Design and Characterization of Materials; Springer Science & Business Media: Berlin/Heidelberg, Germany; New York, NY, USA, 2006; Volume 84, ISBN 978-3-540-31297-0. [Google Scholar]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833. [Google Scholar] [CrossRef] [Green Version]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. II. Overlap Populations, Bond Orders, and Covalent Bond Energies. J. Chem. Phys. 1955, 23, 1841. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO-MO Molecular Wave Functions. III. Effects of Hybridization on Overlap and Gross AO Populations. J. Chem. Phys. 1955, 23, 2338. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO-MO Molecular Wave Functions. IV. Bonding and Antibonding in LCAO and Valence-Bond Theories. J. Chem. Phys. 1955, 23, 2343. [Google Scholar] [CrossRef]
- Ishii, T.; Tsuboi, S.; Sakane, G.; Yamashita, M.; Breedlove, B.K. Universal spectrochemical series of six-coordinate octahedral metal complexes for modifying the ligand field splitting. Dalton Trans. 2009, 4, 680–687. [Google Scholar] [CrossRef]
- Yuan, G.; Lu, P.; Han, L.; Yu, Z.; Shen, Y.; Zhao, L.; Liu, Y. Structural and electronic properties of neutral clusters Ga12X (X = C, Si, Ge, Sn, and Pb) and their anions from first principles. Phys. B Condens. 2011, 406, 3498–3501. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishimura, T.; Toba, T.; Sakane, G.; Ishii, T. Periodicity of Superatomic Hybrid Orbitals in Substituted Superatoms and Superatomic-like X@Ga12 (X = Li~Kr) Clusters. Crystals 2022, 12, 543. https://doi.org/10.3390/cryst12040543
Nishimura T, Toba T, Sakane G, Ishii T. Periodicity of Superatomic Hybrid Orbitals in Substituted Superatoms and Superatomic-like X@Ga12 (X = Li~Kr) Clusters. Crystals. 2022; 12(4):543. https://doi.org/10.3390/cryst12040543
Chicago/Turabian StyleNishimura, Takaki, Teruyuki Toba, Genta Sakane, and Tomohiko Ishii. 2022. "Periodicity of Superatomic Hybrid Orbitals in Substituted Superatoms and Superatomic-like X@Ga12 (X = Li~Kr) Clusters" Crystals 12, no. 4: 543. https://doi.org/10.3390/cryst12040543
APA StyleNishimura, T., Toba, T., Sakane, G., & Ishii, T. (2022). Periodicity of Superatomic Hybrid Orbitals in Substituted Superatoms and Superatomic-like X@Ga12 (X = Li~Kr) Clusters. Crystals, 12(4), 543. https://doi.org/10.3390/cryst12040543