
Towards the Development of Novel Diclofenac Multicomponent Pharmaceutical Solids

 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
Synthesis of Diclofenac Acid Form
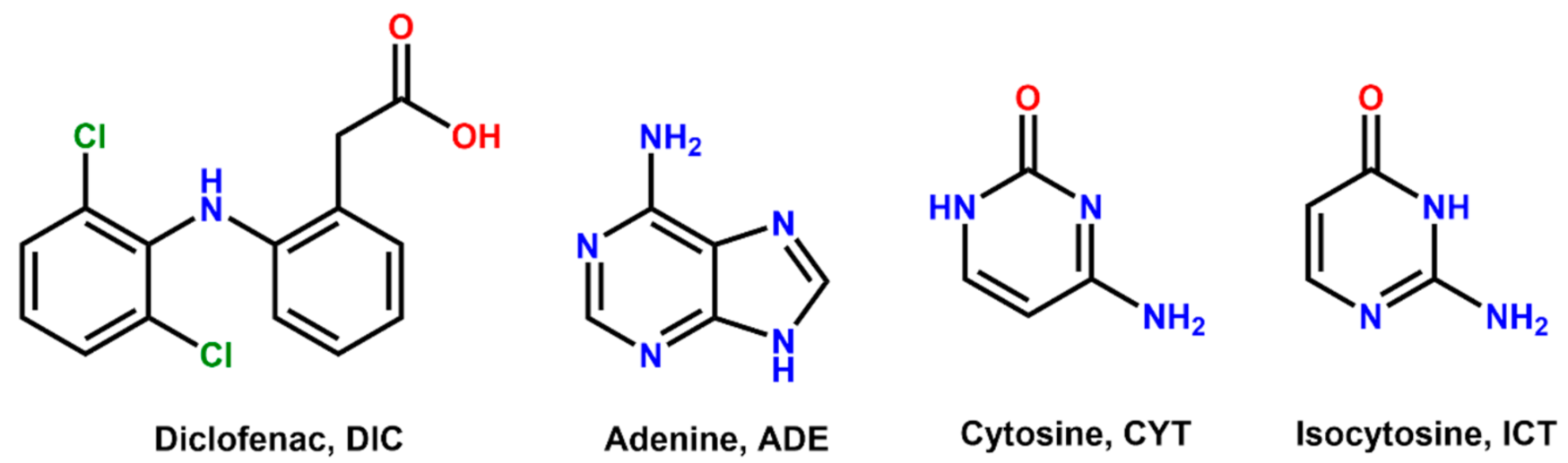
2.2. Coformer Selection
2.3. General Procedure for Mechanochemical Synthesis
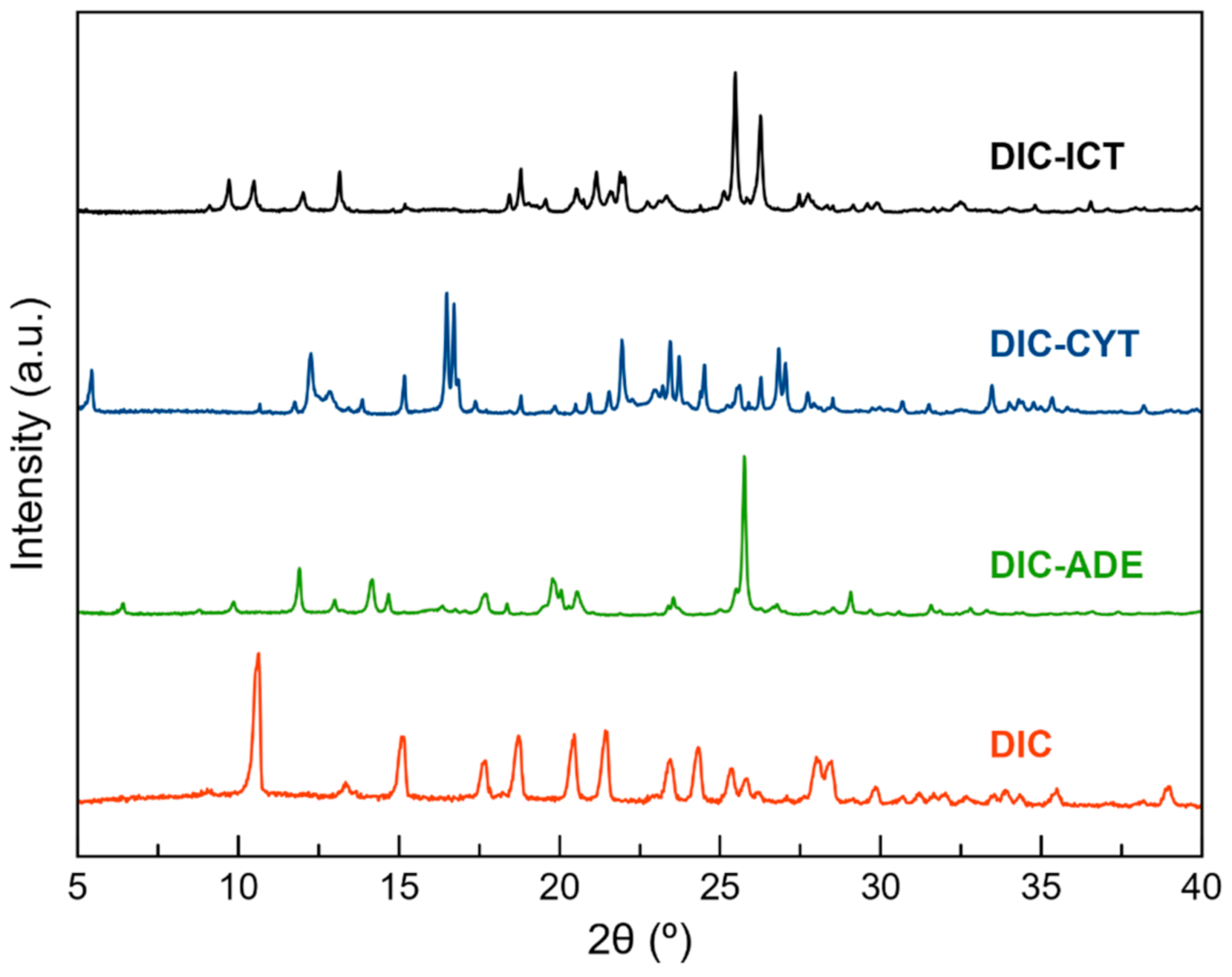
2.4. Powder X-ray Diffraction (PXRD)
2.5. Preparation of Single Crystals
2.6. Single-Crystal X-ray Diffraction (SCXRD)
2.7. Thermal Analysis
2.8. Infrared Spectroscopy
2.9. Stability Test
2.10. Solubility Test
3. Results and Discussion
3.1. Coformer Selection
3.2. Mechanochemical Synthesis
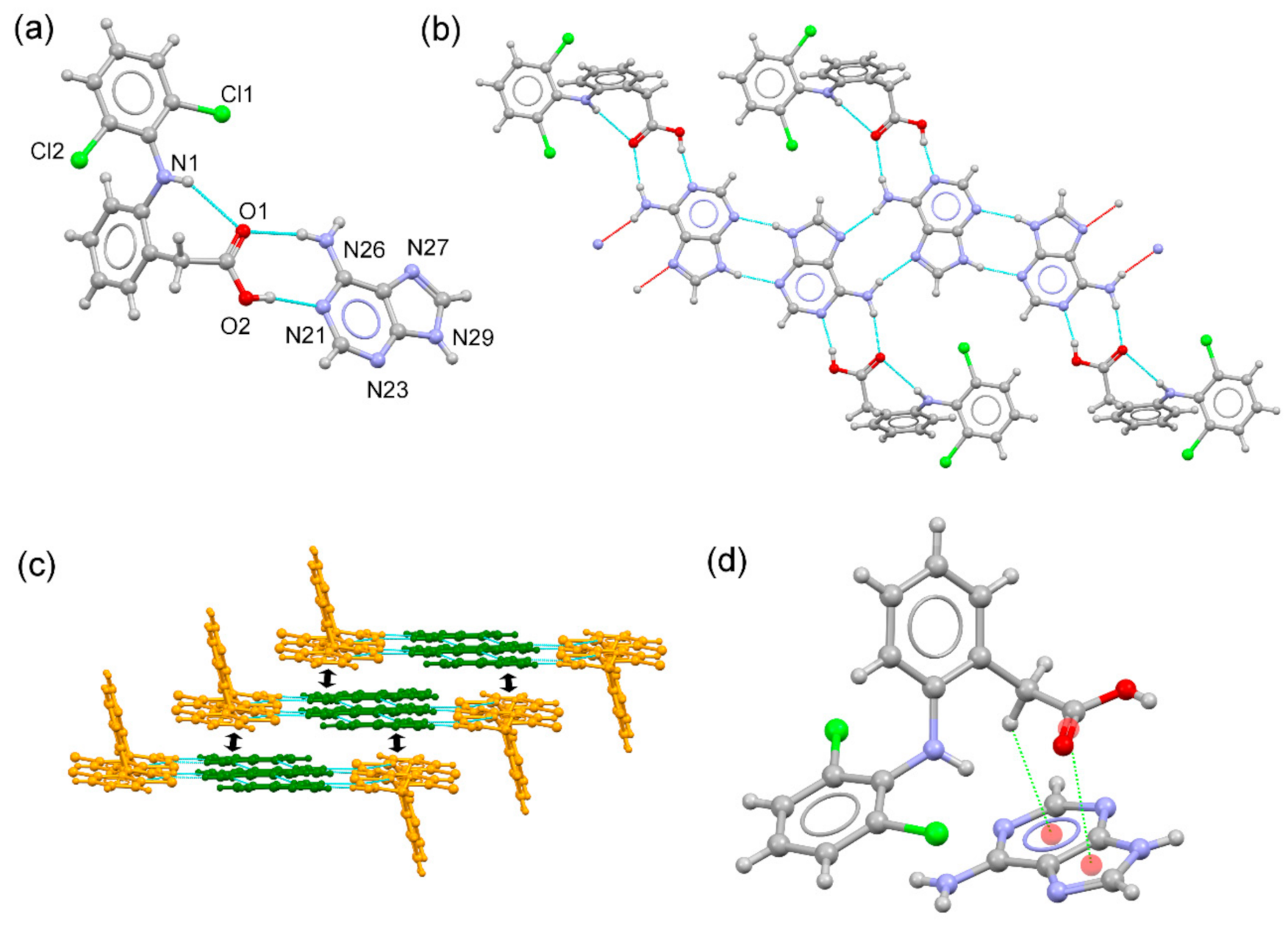
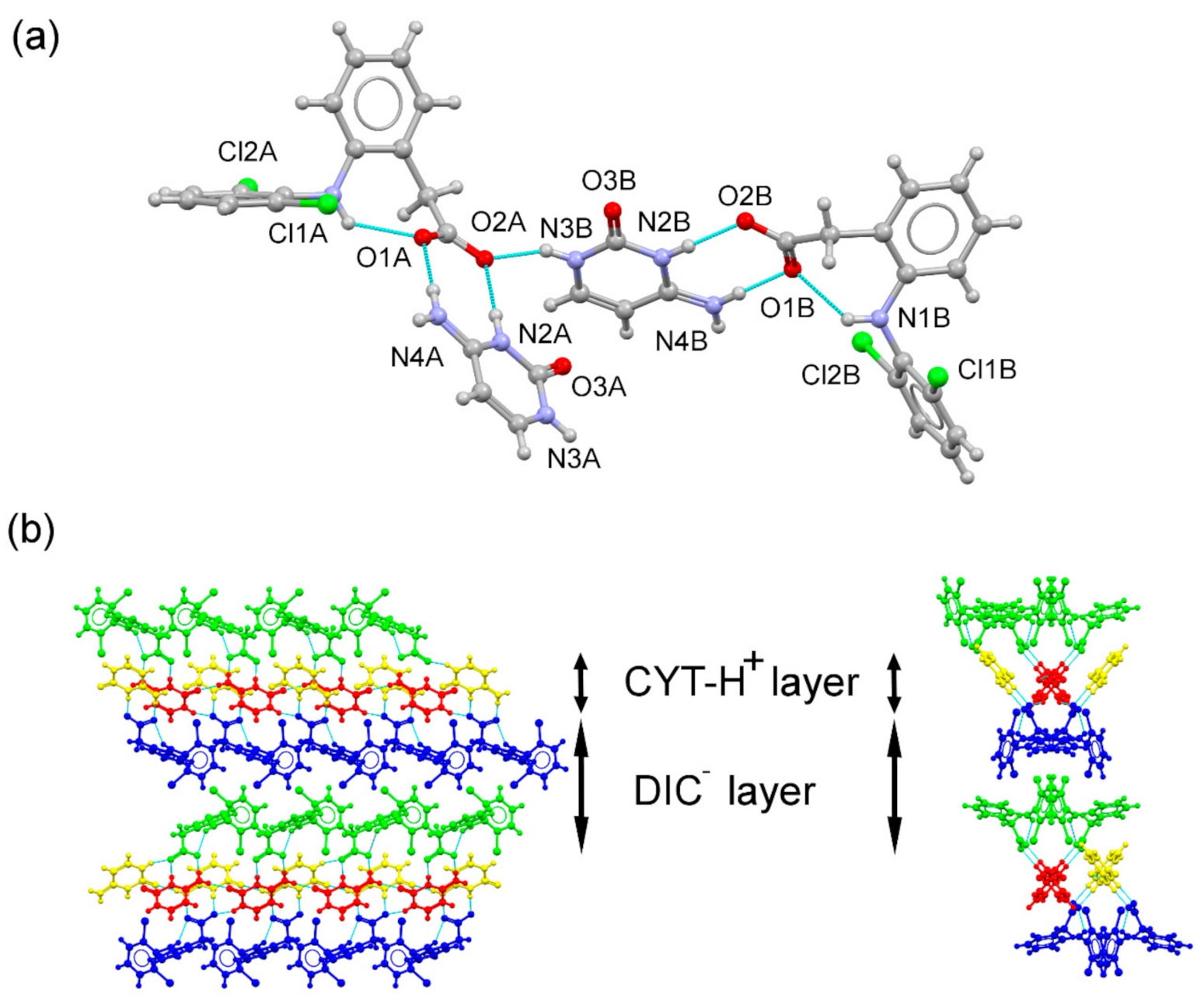
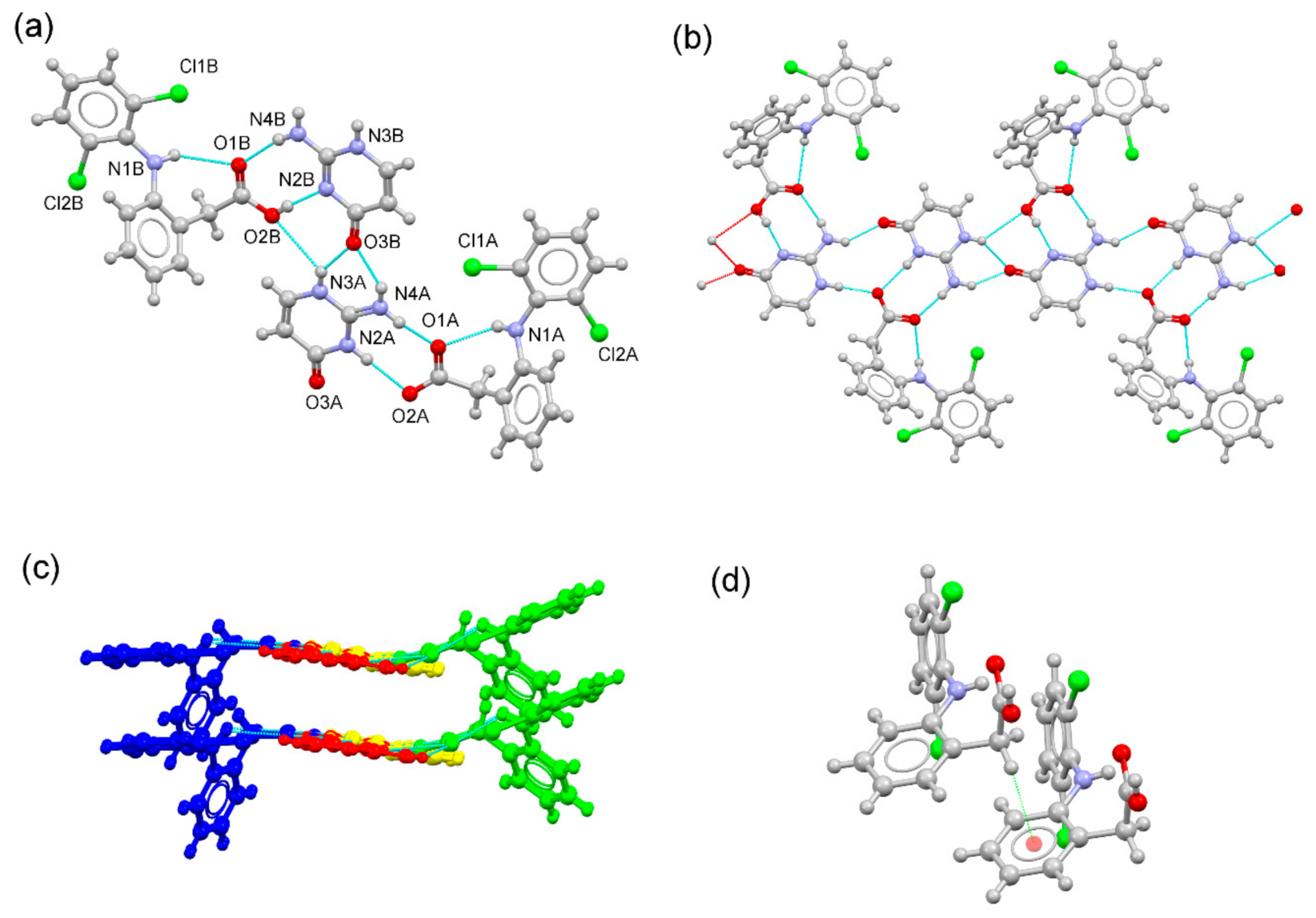
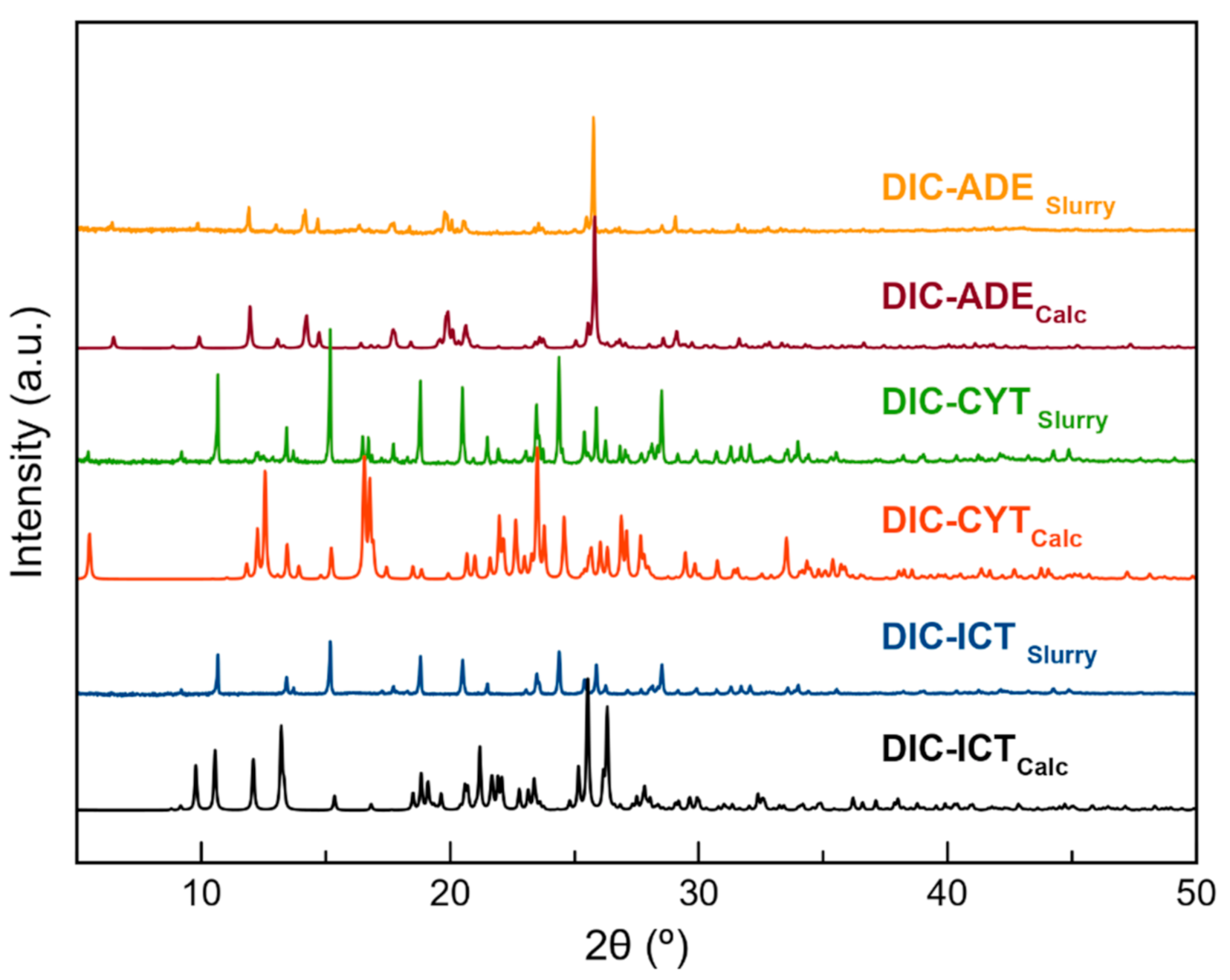
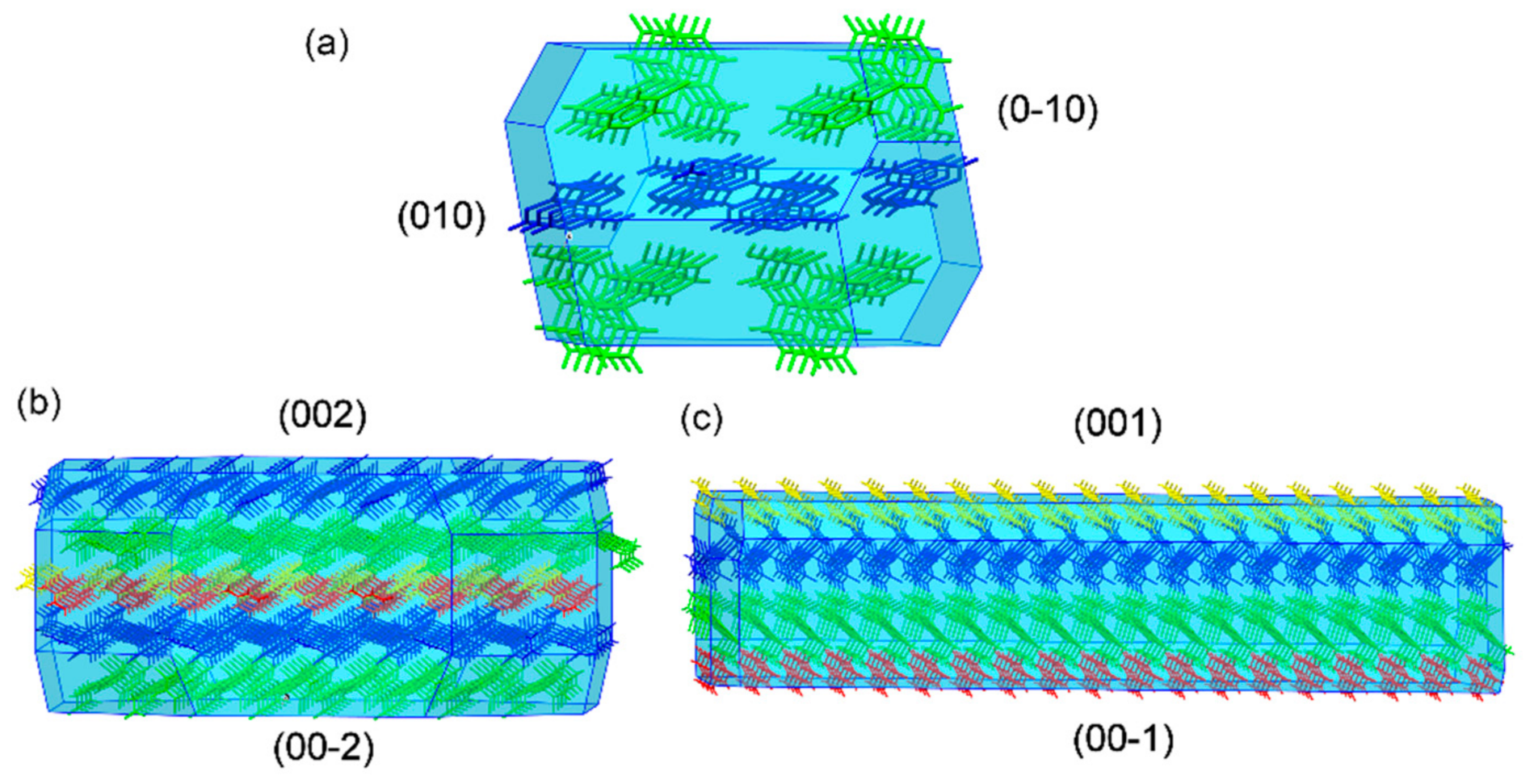
3.3. Structural Analysis of Multicomponent Forms
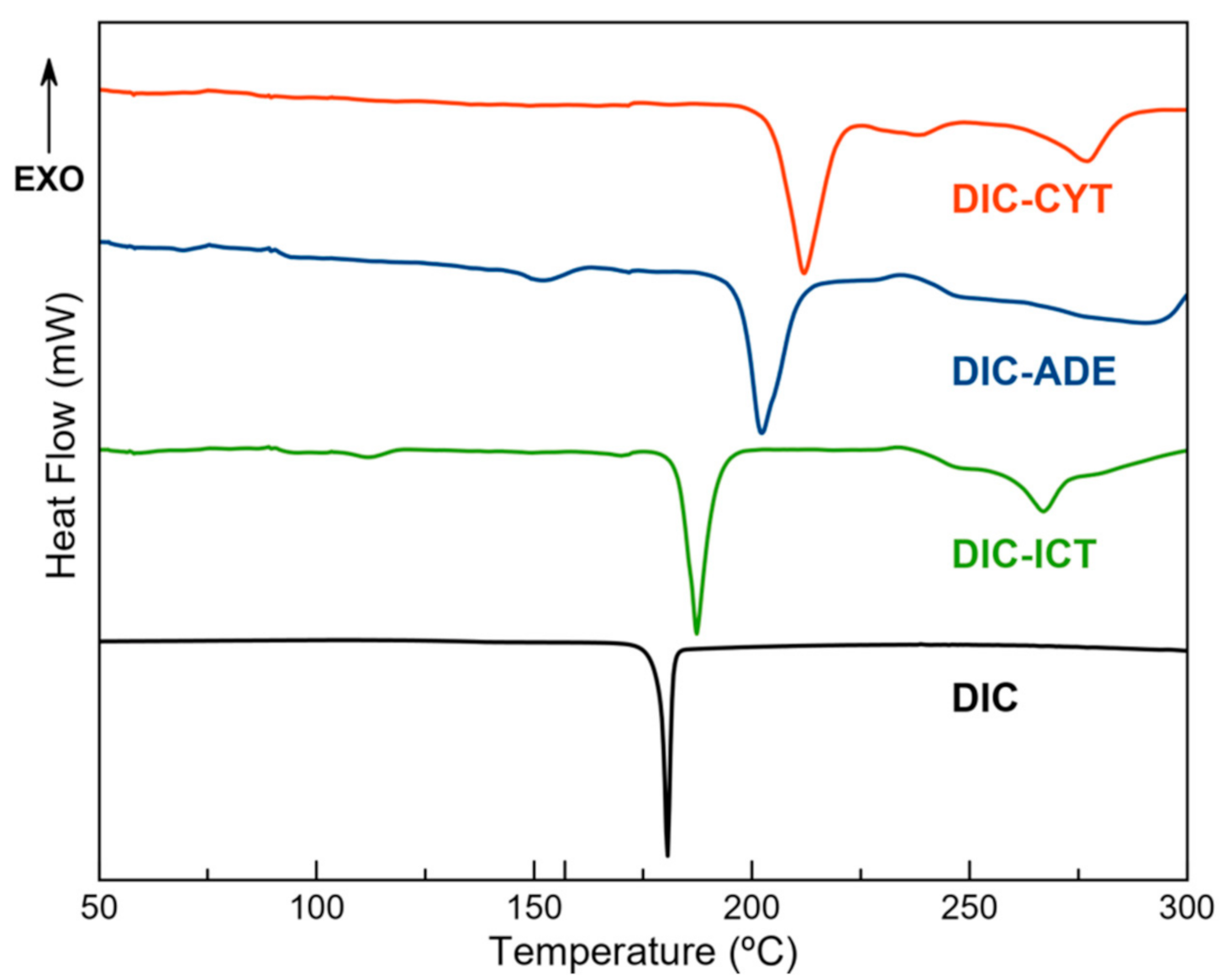
3.4. Thermal Analysis
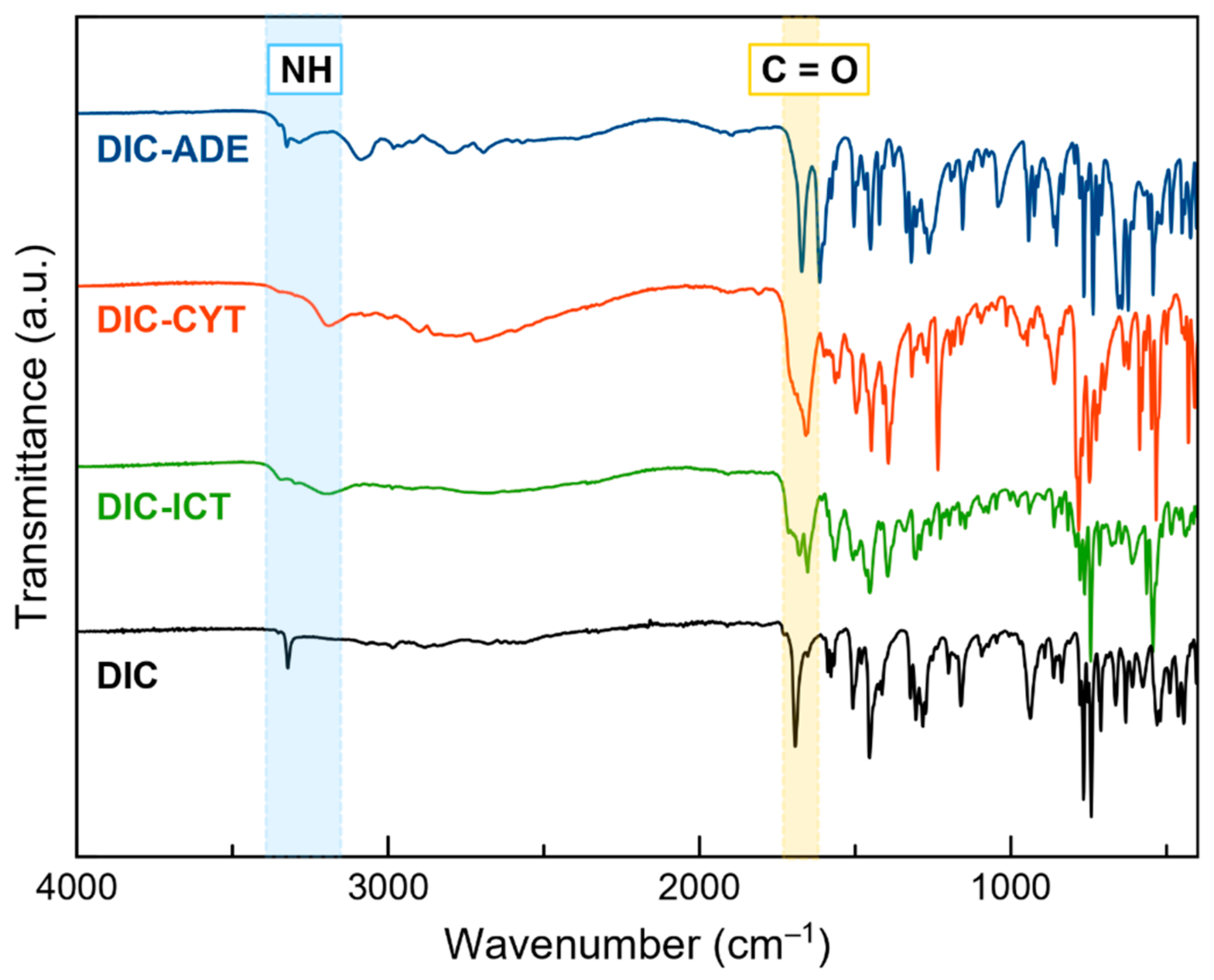
3.5. Fourier Transform Infrared (FT–IR) Spectroscopy
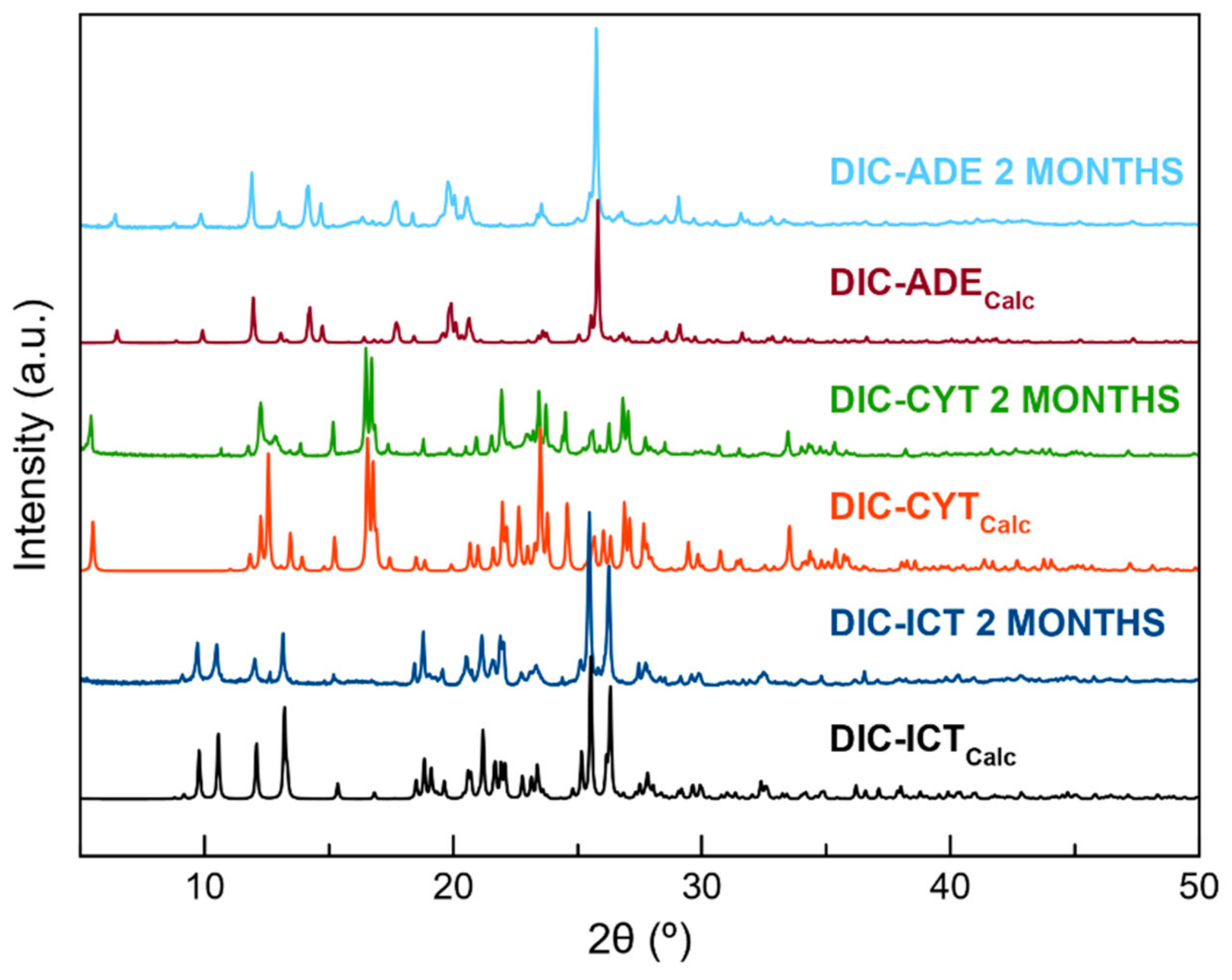
3.6. Stability Studies
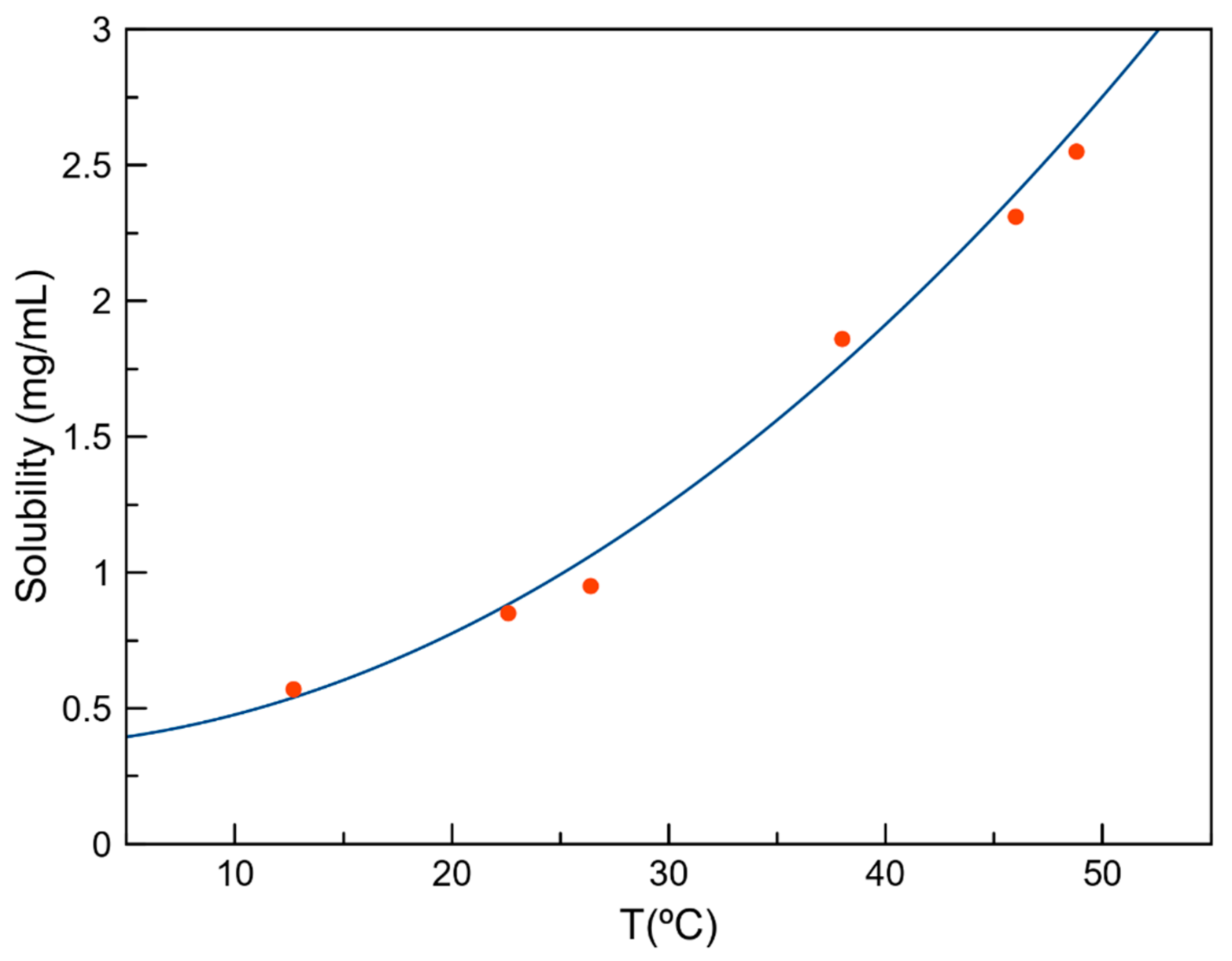
3.7. Solubility Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davies, N.M.; Andersen, K.E. Clinical Pharmacokinetics of Diclofenac. Clin. Pharmacokinet. 2012, 33, 184–213. [Google Scholar] [CrossRef] [PubMed]
- Brogden, R.N.; Heel, R.C.; Pakes, G.E.; Speight, T.M.; Avery, G.S. Diclofenac Sodium: A Review of Its Pharmacological Properties and Therapeutic Use in Rheumatic Diseases and Pain of Varying Origin. Drugs 2012, 20, 24–48. [Google Scholar] [CrossRef] [PubMed]
- Gan, T.J. Diclofenac: An Update on Its Mechanism of Action and Safety Profile. Curr. Med. Res. Opin. 2010, 26, 1715–1731. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A Provisional Biopharmaceutical Classification of the Top 200 Oral Drug Products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef]
- Stuart, M.; Box, K. Chasing Equilibrium: Measuring the Intrinsic Solubility of Weak Acids and Bases. Anal. Chem. 2005, 77, 983–990. [Google Scholar] [CrossRef]
- Gomaa, S. Adverse Effects Induced by Diclofenac, Ibuprofen, and Paracetamol Toxicity on Immunological and Biochemical Parameters in Swiss Albino Mice. J. Basic Appl. Zool. 2018, 79, 5. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.J.; Steed, J.W. Pharmaceutical Cocrystals, Salts and Multicomponent Systems; Intermolecular Interactions and Property Based Design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Bolla, G.; Nangia, A. Pharmaceutical Cocrystals: Walking the Talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef]
- Báthori, N.B.; Lemmerer, A.; Venter, G.A.; Bourne, S.A.; Caira, M.R. Pharmaceutical Co-Crystals with Isonicotinamide-Vitamin B3, Clofibric Acid, and Diclofenac-and Two Isonicotinamide Hydrates. Cryst. Growth Des. 2011, 11, 75–87. [Google Scholar] [CrossRef]
- Feng, W.Q.; Wang, L.Y.; Gao, J.; Zhao, M.Y.; Li, Y.T.; Wu, Z.Y.; Yan, C.W. Solid State and Solubility Study of a Potential Anticancer Drug-Drug Molecular Salt of Diclofenac and Metformin. J. Mol. Struct. 2021, 1234, 130166. [Google Scholar] [CrossRef]
- Nugrahani, I.; Utami, D.; Ibrahim, S.; Nugraha, Y.P.; Uekusa, H. Zwitterionic Cocrystal of Diclofenac and L-Proline: Structure Determination, Solubility, Kinetics of Cocrystallization, and Stability Study. Eur. J. Pharm. Sci. 2018, 117, 168–176. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Manin, A.N.; Manin, N.G.; Kuzmina, L.G.; Churakov, A.V.; Perlovich, G.L. Pharmaceutical Cocrystals of Diflunisal and Diclofenac with Theophylline. Mol. Pharm. 2014, 11, 3707–3715. [Google Scholar] [CrossRef]
- Goswami, P.K.; Kumar, V.; Ramanan, A. Multicomponent Solids of Diclofenac with Pyridine Based Coformers. J. Mol. Struct. 2020, 1210, 128066. [Google Scholar] [CrossRef]
- Sivakova, S.; Rowan, S.J. Nucleobases as Supramolecular Motifs. Chem. Soc. Rev. 2005, 34, 9–21. [Google Scholar] [CrossRef]
- Koch, E.S.; McKenna, K.A.; Kim, H.J.; Young, V.G.; Swift, J.A. Thymine Cocrystals Based on DNA-Inspired Binding Motifs. CrystEngComm 2017, 19, 5679–5685. [Google Scholar] [CrossRef]
- Sarai, A.; Kono, H. Protein-DNA Recognition Patterns and Predictions. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 379–398. [Google Scholar] [CrossRef]
- Xia, Y.; Qu, F.; Peng, L. Triazole Nucleoside Derivatives Bearing Aryl Functionalities on the Nucleobases Show Antiviral and Anticancer Activity. Mini-Rev. Med. Chem. 2010, 10, 806–821. [Google Scholar] [CrossRef]
- Sun, R.; Wang, L. Inhibition of Mycoplasma Pneumoniae Growth by FDA-Approved Anticancer and Antiviral Nucleoside and Nucleobase Analogs. BMC Microbiol. 2013, 13, 184. [Google Scholar] [CrossRef] [Green Version]
- Allen, F.H. The Cambridge Structural Database: A Quarter of a Million Crystal Structures and Rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Loschen, C.; Klamt, A. Solubility Prediction, Solvate and Cocrystal Screening as Tools for Rational Crystal Engineering. J. Pharm. Pharmacol. 2015, 67, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Bruker APEX4, APEX4 V2021.1 2021; Bruker-AXS: Madison, WI, USA, 2021.
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Castellari, C.; Ottani, S. Two Monoclinic Forms of Diclofenac Acid. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1997, 53, 794–797. [Google Scholar] [CrossRef]
- Horst, J.H.T.; Deij, M.A.; Cains, P.W. Discovering New Co-Crystals. Cryst. Growth Des. 2009, 9, 1531–1537. [Google Scholar] [CrossRef]
- Moser, P.; Sallmann, A.; Wiesenberg, I. Synthesis and Quantitative Structure-Activity Relationships of Diclofenac Analogues. J. Med. Chem. 1990, 33, 2358–2368. [Google Scholar] [CrossRef]
- Jaiboon, N.; Yos-In, K.; Ruangchaithaweesuk, S.; Chaichit, N.; Thutivoranath, R.; Siritaedmukul, K.; Hannongbua, S. New Orthorhombic Form of 2-[(2,6-Dichlorophenyl)Amino]Benzeneacetic Acid (Diclofenac Acid). Anal. Sci. 2001, 17, 1465–1466. [Google Scholar] [CrossRef] [Green Version]
- Hamamci Alisir, S.; Dege, N. Crystal Structure of a Mixed-Ligand Silver(I) Complex of the Non-Steroidal Anti-Inflammatory Drug Diclofenac and Pyrimidine. Acta Crystallogr. Sect. E Crystallogr. Commun. 2016, 72, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Caglar, S.; Dilek, E.; Caglar, B.; Adiguzel, E.; Temel, E.; Buyukgungor, O.; Tabak, A. New Metal Complexes with Diclofenac Containing 2-Pyridineethanol or 2-Pyridinepropanol: Synthesis, Structural, Spectroscopic, Thermal Properties, Catechol Oxidase and Carbonic Anhydrase Activities. J. Coord. Chem. 2016, 69, 3321–3335. [Google Scholar] [CrossRef]
- García-García, A.; Méndez-Arriaga, J.M.; Martín-Escolano, R.; Cepeda, J.; Gómez-Ruiz, S.; Salinas-Castillo, A.; Seco, J.M.; Sánchez-Moreno, M.; Choquesillo-Lazarte, D.; Ruiz-Muelle, A.B.; et al. In Vitro Evaluation of Leishmanicidal Properties of a New Family of Monodimensional Coordination Polymers Based on Diclofenac Ligand. Polyhedron 2020, 184, 114570. [Google Scholar] [CrossRef]
- Sharma, R.; Sharma, R.P.; Bala, R.; Kariuki, B.M. Second Sphere Coordination in Oxoanion Binding: Synthesis, Spectroscopic Characterisation and Crystal Structures of Trans-[Bis(Ethylenediamine)Dinitrocobalt(III)] Diclofenac and Chlorate. J. Mol. Struct. 2007, 826, 177–184. [Google Scholar] [CrossRef]
- Biswas, P.; Dastidar, P. Anchoring Drugs to a Zinc(II) Coordination Polymer Network: Exploiting Structural Rationale toward the Design of Metallogels for Drug-Delivery Applications. Inorg. Chem. 2021, 60, 3218–3231. [Google Scholar] [CrossRef]
- Lu, C.; Laws, K.; Eskandari, A.; Suntharalingam, K. A Reactive Oxygen Species-Generating, Cyclooxygenase-2 Inhibiting, Cancer Stem Cell-Potent Tetranuclear Copper(II) Cluster. Dalton Trans. 2017, 46, 12785–12789. [Google Scholar] [CrossRef] [Green Version]
- Sayen, S.; Carlier, A.; Tarpin, M.; Guillon, E. A Novel Copper(II) Mononuclear Complex with the Non-Steroidal Anti-Inflammatory Drug Diclofenac: Structural Characterization and Biological Activity. J. Inorg. Biochem. 2013, 120, 39–43. [Google Scholar] [CrossRef]
- Lee, S.; Kapustin, E.A.; Yaghi, O.M. Coordinative Alignment of Molecules in Chiral Metal-Organic Frameworks. Science 2016, 353, 808–811. [Google Scholar] [CrossRef]
- Paul, M.; Sarkar, K.; Deb, J.; Dastidar, P. Hand-Ground Nanoscale ZnII-Based Coordination Polymers Derived from NSAIDs: Cell Migration Inhibition of Human Breast Cancer Cells. Chem.—A Eur. J. 2017, 23, 5736–5747. [Google Scholar] [CrossRef]
- Dilek, E.; Caglar, S.; Erdogan, K.; Caglar, B.; Sahin, O. Synthesis and Characterization of Four Novel Palladium(II) and Platinum(II) Complexes with 1-(2-Aminoethyl)Pyrrolidine, Diclofenac and Mefenamic Acid: In Vitro Effect of These Complexes on Human Serum Paraoxanase1 Activity. J. Biochem. Mol. Toxicol. 2018, 32, e22043. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Grepioni, F. Mechanochemical Preparation of Co-Crystals. Chem. Soc. Rev. 2013, 42, 7638–7648. [Google Scholar] [CrossRef]
- Karki, S.; Friščić, T.; Jones, W.; Motherwell, W.D.S. Screening for Pharmaceutical Cocrystal Hydrates via Neat and Liquid-Assisted Grinding. Mol. Pharm. 2007, 4, 347–354. [Google Scholar] [CrossRef]
- Perumalla, S.R.; Pedireddi, V.R.; Sun, C.C. Protonation of Cytosine: Cytosinium vs. Hemicytosinium Duplexes. Cryst. Growth Des. 2013, 13, 429–432. [Google Scholar] [CrossRef]
- Childs, S.L.; Stahly, G.P.; Park, A. The Salt-Cocrystal Continuum: The Influence of Crystal Structure on Ionization State. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef] [Green Version]
- Perlovich, G. Melting Points of One- and Two-Component Molecular Crystals as Effective Characteristics for Rational Design of Pharmaceutical Systems. Acta Cryst. B Struct. Sci. Cryst. Eng. Mater. 2020, 76, 696–706. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Chakraborty, S.; Ganguly, S.; Desiraju, G.R. Synthon Identification in Co-Crystals and Polymorphs with IR Spectroscopy. Primary Amides as a Case Study. CrystEngComm 2013, 15, 4640–4654. [Google Scholar] [CrossRef]
- Heinz, A.; Strachan, C.J.; Gordon, K.C.; Rades, T. Analysis of Solid-State Transformations of Pharmaceutical Compounds Using Vibrational Spectroscopy. J. Pharm. Pharmacol. 2009, 61, 971–988. [Google Scholar] [CrossRef]
- Glomme, A.; März, J.; Dressman, J.B. Comparison of a Miniaturized Shake-Flask Solubility Method with Automated Potentiometric Acid/Base Titrations and Calculated Solubilities. J. Pharm. Sci. 2005, 94, 1–16. [Google Scholar] [CrossRef]
- Nugrahani, I.; Kumalasari, R.A.; Auli, W.N.; Horikawa, A.; Uekusa, H. Salt Cocrystal of Diclofenac Sodium-l-Proline: Structural, Pseudopolymorphism, and Pharmaceutics Performance Study. Pharmaceutics 2020, 12, 690. [Google Scholar] [CrossRef]
- Putra, O.D.; Umeda, D.; Nugraha, Y.P.; Furuishi, T.; Nagase, H.; Fukuzawa, K.; Uekusa, H.; Yonemochi, E. Solubility Improvement of Epalrestat by Layered Structure Formation: Via Cocrystallization. CrystEngComm 2017, 19, 2614–2622. [Google Scholar] [CrossRef]
- Putra, O.D.; Umeda, D.; Fujita, E.; Haraguchi, T.; Uchida, T.; Yonemochi, E.; Uekusa, H. Solubility Improvement of Benexate through Salt Formation Using Artificial Sweetener. Pharmaceutics 2018, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Sanphui, P.; Rajput, L. Tuning Solubility and Stability of Hydrochloro-Thiazide Co-Crystals. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 81–90. [Google Scholar] [CrossRef]
- McNamara, D.P.; Childs, S.L.; Giordano, J.; Iarriccio, A.; Cassidy, J.; Shet, M.S.; Mannion, R.; O’Donnell, E.; Park, A. Use of a Glutaric Acid Cocrystal to Improve Oral Bioavailability of a Low Solubility API. Pharm. Res. 2006, 23, 1888–1897. [Google Scholar] [CrossRef]
- Childs, S.L.; Chyall, L.J.; Dunlap, J.T.; Smolenskaya, V.N.; Stahly, B.C.; Stahly, G.P. Crystal Engineering Approach to Forming Cocrystals of Amine Hydrochlorides with Organic Acids. Molecular Complexes of Fluoxetine Hydrochloride with Benzoic, Succinic, and Fumaric Acids. J. Am. Chem. Soc. 2004, 126, 13335–13342. [Google Scholar] [CrossRef] [Green Version]
- Sathisaran, I.; Dalvi, S. Engineering Cocrystals of Poorly Water-Soluble Drugs to Enhance Dissolution in Aqueous Medium. Pharmaceutics 2018, 10, 108. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | DIC form I * | DIC form II * | DIC–ADE | DIC–CYT | DIC–ICT |
| Formula | C14H11Cl2NO2 | C14H11Cl2NO2 | C19H16Cl2N6O2 | C36H32Cl4N8O6 | C36H32Cl4N8O6 |
| Formula weight | 296.14 | 296.14 | 431.28 | 814.49 | 814.49 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Orthorhombic | Triclinic |
| Space group | C2/c | P21/c | P-1 | Pca21 | P1 |
| a/Å | 20.226 (4) | 8.384 (2) | 7.0545 (2) | 13.8431 (4) | 4.720 (2) |
| b/Å | 6.971 (3) | 10.898 (2) | 10.3452 (4) | 8.4502 (4) | 9.701 (3) |
| c/Å | 20.061 (4) | 14.822 (5) | 14.3310 (5) | 32.0448 (11) | 20.189 (7) |
| α/° | 90 | 90 | 97.913 (2) | 90 | 84.328 (16) |
| β/° | 109.64 (2) | 92.76 (2) | 104.237 (2) | 90 | 88.058 (16) |
| γ/° | 90 | 90 | 100.934 (2) | 90 | 85.963 (16) |
| V/Å3 | 2664 (1) | 1352.7 (6) | 976.51 (6) | 3748.5 (2) | 917.4 (6) |
| Z | 8 | 4 | 2 | 4 | 1 |
| Dc/g cm−3 | 1.477 | 1.454 | 1.467 | 1.443 | 1.474 |
| F(000) | 1216 | 608 | 444 | 1680 | 420 |
| Reflections collected | 4383 | 4079 | 12246 | 29559 | 6125 |
| Unique reflections | 2589 | 3940 | 3398 | 6581 | 6125 |
| Data/restraints/parameters | 2582/36/217 | 3937/36/216 | 3398/0/263 | 6581/1/487 | 6125/3/488 |
| Goodness-of-fit (on F2) | 1.057 | 1.005 | 1.066 | 1.016 | 1.059 |
| R1 [I > 2σ(I)] | 0.0374 | 0.0397 | 0.0526 | 0.0506 | 0.0683 |
| wR2 [I > 2σ(I)] | 0.0992 | 0.0859 | 0.1531 | 0.1302 | 0.1800 |
| Absolute structure parameter | - | - | - | 0.067 (15) | 0.01 (2) |
| CCDC | 128772 | 128771 | 2180776 | 2180777 | 2180778 |
| Coformer | Hex (kcal/mol) | |
|---|---|---|
| Glycine | −5.070 | |
| Proline | −4.743 | Ref. [11] |
| Alanine | −3.949 | |
| Glutamic Acid | −3.699 | |
| Aspartic Acid | −3.285 | |
| Cytosine | −3.177 | This work |
| Adenine | −2.393 | This work |
| Cysteine | −2.015 | |
| Thymine | −1.498 | |
| Phenylglycine | −1.09 | |
| Isocytosine | −1.075 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acebedo-Martínez, F.J.; Alarcón-Payer, C.; Barrales-Ruiz, H.M.; Niclós-Gutiérrez, J.; Domínguez-Martín, A.; Choquesillo-Lazarte, D. Towards the Development of Novel Diclofenac Multicomponent Pharmaceutical Solids. Crystals 2022, 12, 1038. https://doi.org/10.3390/cryst12081038
Acebedo-Martínez FJ, Alarcón-Payer C, Barrales-Ruiz HM, Niclós-Gutiérrez J, Domínguez-Martín A, Choquesillo-Lazarte D. Towards the Development of Novel Diclofenac Multicomponent Pharmaceutical Solids. Crystals. 2022; 12(8):1038. https://doi.org/10.3390/cryst12081038
Chicago/Turabian StyleAcebedo-Martínez, Francisco Javier, Carolina Alarcón-Payer, Helena María Barrales-Ruiz, Juan Niclós-Gutiérrez, Alicia Domínguez-Martín, and Duane Choquesillo-Lazarte. 2022. "Towards the Development of Novel Diclofenac Multicomponent Pharmaceutical Solids" Crystals 12, no. 8: 1038. https://doi.org/10.3390/cryst12081038
APA StyleAcebedo-Martínez, F. J., Alarcón-Payer, C., Barrales-Ruiz, H. M., Niclós-Gutiérrez, J., Domínguez-Martín, A., & Choquesillo-Lazarte, D. (2022). Towards the Development of Novel Diclofenac Multicomponent Pharmaceutical Solids. Crystals, 12(8), 1038. https://doi.org/10.3390/cryst12081038