1. Introduction
In the past years, crystal engineering and, more particularly, cocrystal design has raised the attention of the pharmaceutical industry as an efficient method to develop new pharmaceutical solid forms. The main advantages of such pharmaceutical cocrystals include not only the enhancement of the physicochemical properties of active pharmaceutical ingredients (APIs) but also the potential of obtaining synergic effects in codrug formulations, keeping the options for intellectual property rights open at lower costs.
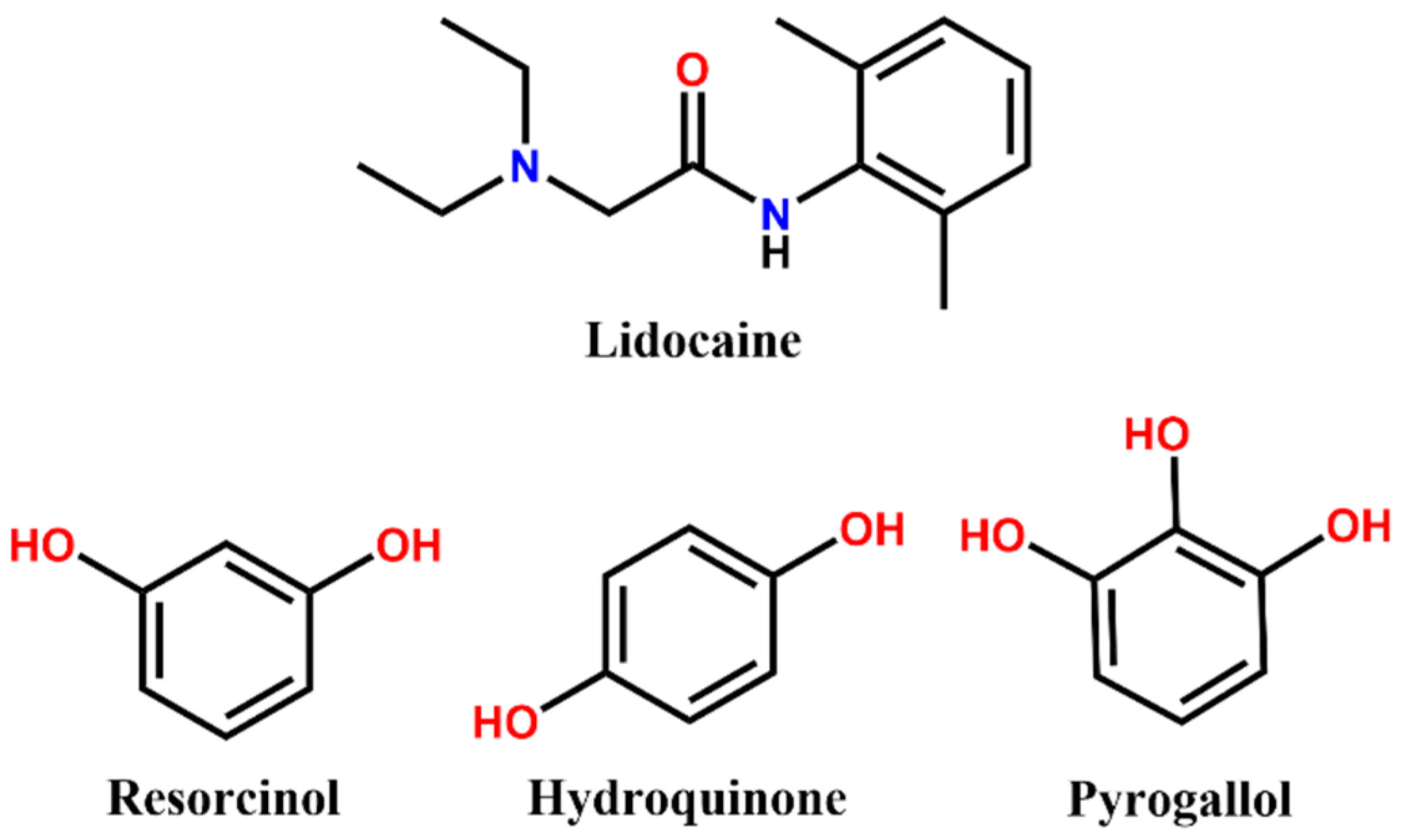
Lidocaine (2-diethylamino-
N-(2,6-dimethylphenyl)acetamide), hereafter
(lid), is an active pharmaceutical ingredient widely used as an anaesthetic in intravenous injection to treat and prevent pain [
1,
2] in some medical procedures. It is also used in clinics as an antiarrhythmic drug [
3] to treat ventricular arrhythmias, specifically ventricular tachycardia and ventricular fibrillation, or as a vasoconstrictor in topical applications [
4].
(Lid) shows low solubility in the base form [
5]. Therefore, in pharmaceutical formulations, lidocaine is generally used as its hydrochloride derivative
(lidhcl). Solubility problems are certainly a big concern regarding the efficacy of oral administration drugs. Hence, if
(lid) wants to be directly included in drug formulations, one of the best approaches seems to be the development of novel multicomponent pharmaceutical solids, a well-established method able to modulate the physicochemical and biopharmaceutical properties of APIs [
6], such as stability, solubility, or manufacturability. In this context, only a few studies can be found in the literature reporting a lidocaine base [
7,
8,
9], where
(lid) salts with improved properties were obtained. To build such multicomponent solids, the lidocaine molecule offers different functional groups able to participate in supramolecular synthons (
Figure 1), e.g., an amide group that allows hydrogen bonding and an aromatic moiety that can interact through π interactions. These groups are good candidates for interaction with other aromatic rings and alcohol groups, among others.
Polyhydroxy benzenes (
Figure 1) are a group of compounds intensely studied and utilised as coformers with multiple APIs including lidocaine [
7]. Indeed, they are included in the Generally Recognised as Safe (GRAS) or Substances Added to Food (EAFUS) lists of the US Food and Drug Administration (FDA). Interestingly, these compounds are quite good H-donors; thus, they can easily form hydrogen bonds with other H-acceptor groups such as amide groups, making them excellent candidates to act as coformers in lidocaine formulations, as already reported for phloroglucinol [
7], allowing a comparative study of the structural effect of chlorine in the final products [
7].
In this work, we focus on comparing the ability of
(lid) and
(lidhcl) to cocrystallise with hydroquinone, resorcinol, and pyrogallol, to form multicomponent pharmaceutical solids, and we study how structure can affect physicochemical properties and usability compared to the parent API. The reported multicomponent solids of
(lid) and
(lidhcl) were cocrystallised through liquid-assisted grinding (LAG), a versatile and green synthetic method for obtaining solid forms [
10,
11] that uses mechanical forces to induce chemical transformations, which is a fast and appropriate tool for multicomponent form screening. The resulting multicomponent forms were characterised by powder X-ray diffraction (PXRD), X-ray differential scanning calorimetry/thermogravimetric analysis (DSC/TGA), Fourier-transform infrared spectroscopy (FTIR), and single-crystal X-ray diffraction (SCXRD). In addition, a thorough analysis of the structural details of the corresponding solids forms obtained for
(lid) and
(lidhcl) was carried out to unravel the influence of the structure on some relevant physicochemical properties, i.e., solubility and stability.
2. Materials and Methods
All compounds were commercially available from Sigma-Aldrich (St. Louis, MO, USA) and used as received. Solvents were of HPLC grade and were also supplied from Sigma-Aldrich.
2.1. Liquid-Assisted Grinding (LAG)
For the LAG [
10,
12,
13] experiments, different molar ratios of about 100 mg scale and 100 µL of dichloromethane (DCM) were added to each tube. Stoichiometric mixtures of lidocaine
(lid) and
(lidhc) with the coformers resorcinol
(res), hydroquinone
(hq), and pyrogallol
(pyr) were gently ground for 30 min at 25 Hz in a Retsch MM400 ball mill (Haan, Germany). A multiple milling homemade accessory allowing for grinding 12 samples at once was used, placing 12 2 mL Eppendorf tubes with three corundum 1 mm balls each.
Three different stoichiometries (1:1, 2:1, 1:2) were screened for each system. The powder materials obtained were analysed by PXRD, FTIR, and DSC/TGA to determine the formation of cocrystals. Cocrystals exhibited distinct PXRD patterns and melting points compared to the starting materials.
2.2. Stability Experiments
In order to investigate the stability with respect to dissociation, a suspension of about 100 mg scale was made with 0.5–1 mL of 0.9% NaCl solution. The suspensions were subjected to magnetic stirring at ambient conditions for 24 h without drying completely, keeping a slurry all the time. Aliquots of the slurry were taken, gently ground, and analysed by PXRD to determine if the cocrystal was dissociated into its components, suffered any transformation, or remained stable in the cocrystal form.
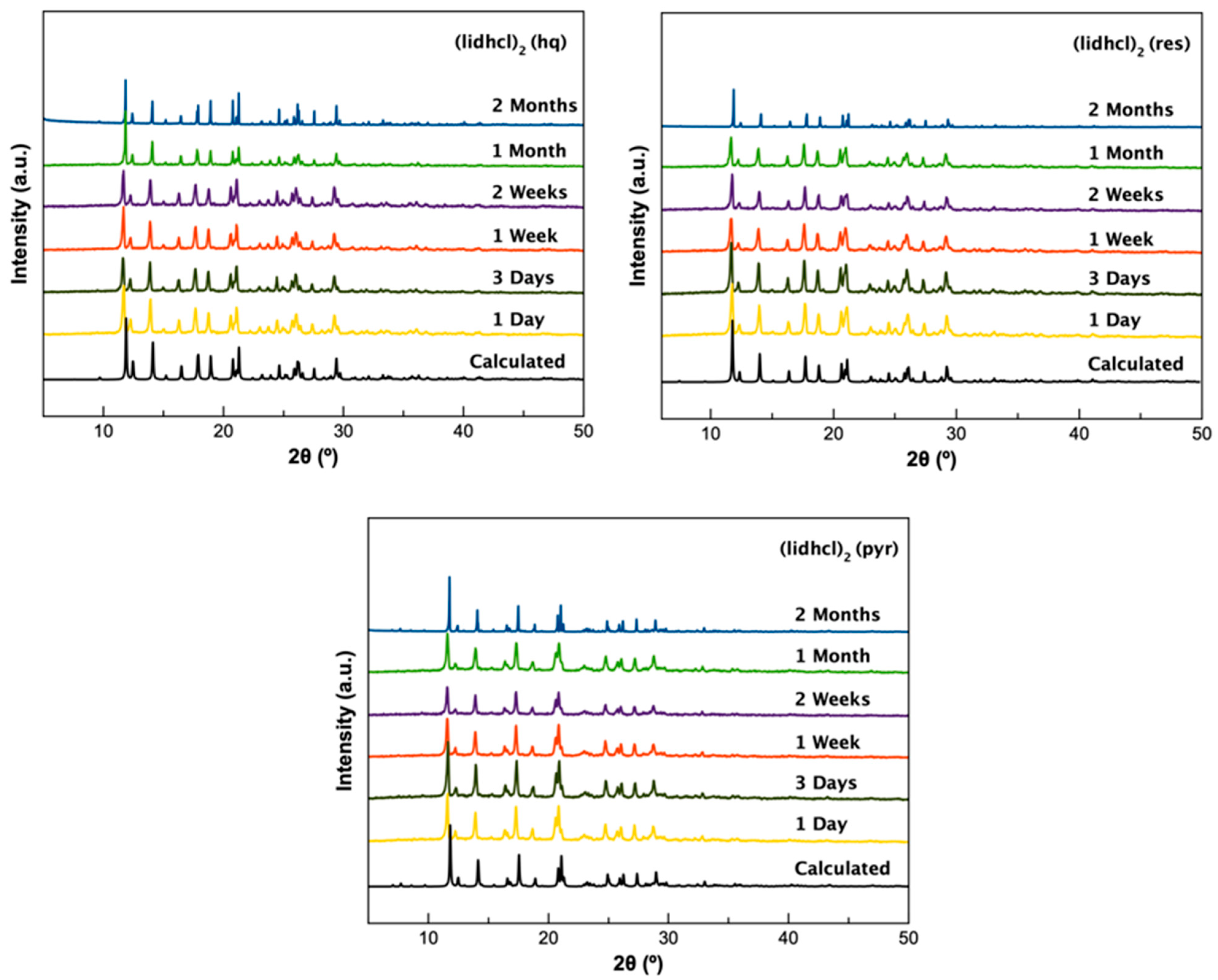
To study the influence of temperature and humidity on the stability of the new phases, these materials were left in a temperature/humidity-controlled chamber with a temperature of 40 °C and 75% relative humidity for 2 months, taking sample aliquots during this time to be analysed by PXRD to evaluate the stability of the crystalline phase. All the samples remained stable as a cocrystal form after 2 months.
2.3. Hetero-Seeding Experiments
In order to obtain single crystals suitable for SCXRD characterisation, evaporation experiments were performed from saturated solutions of the different powders obtained from LAG experiments in DCM. In almost all six cases, single crystals suitable for SCXRD were obtained, except for the (lid)2(res) phase, which only formed a microcrystalline material. A hetero-seeding approach was used to obtain single crystals for this phase, using microcrystalline phases obtained for other coformers with predictably similar structures, getting good results using (lid)2(hq) as the hetero-seed. This was confirmed after the structure solution because both new cocrystals were isostructural.
For the seeding experiment, a few micrometric solid particles of hetero-seed powder (lid)2(hq) were added to the liquified mixture obtained from (lid)2(res) LAG experiments, which immediately started to crystallise as single crystals later identified as (lid)2(res).
For the remaining phases, (lid)2(hq), (lid)2(pyr), (lidhcl)2(hq), (lidhcl)2(res), and (lidhcl)2(pyr) were obtained by direct recrystallisation from the oily liquid obtained in LAG experiments.
2.4. Powder X-ray Diffraction
PXRD patterns were measured on a Bruker D8 Advance Series II Vario diffractometer (Bruker, AXS, Karlsruhe, Germany) using Cu-Kα1 radiation (λ = 1.5406 Å) at 40 kV and 40 mA. Diffraction patterns were collected over 2θ range of 5–60° and using a continuous step size of 0.02° and a total acquisition time of 1 h. The software used for data analysis was Diffrac.EVA v5.0 and TOPAS v6.0 (Bruker, AXS, Karlsruhe, Germany).
2.5. Single-Crystal X-ray Diffraction
Measured crystals were prepared under inert conditions immersed in perfluoropolyether as the protecting oil for manipulation. Suitable crystals were mounted on MiTeGen Micromounts™ (95 Brown Rd, Ithaca, NY, USA) and these samples were used for data collection. Data were collected with a Bruker D8 Venture diffractometer and processed with the APEX3 suite [
14]. Structures were solved by direct methods [
15], which revealed the position of all non-hydrogen atoms. These atoms were refined on F
2 by a full-matrix least-squares procedure using anisotropic displacement parameters [
15]. All hydrogen atoms were located by difference Fourier maps and included as fixed contributions riding on attached atoms with isotropic thermal displacement parameters 1.2 times those of the respective atom. Geometric calculations and molecular graphics were performed with Mercury [
16] and Olex2 [
17]. Additional crystal data are shown in
Table 1.
2.6. Thermal Analysis
For the DSC/TGA experiments, samples in the range of 30 mg were studied using a Mettler Toledo TGA/DSC 3+ Star analyser. Samples were heated at 10 °C/min in the temperature range 25–190 °C under a nitrogen atmosphere with 100 mL/min flow in aluminium capsules.
2.7. Fourier-Transform Infrared Spectra
Fourier-transform infrared spectra (FTIR) were recorded with an attenuated total reflectance (ATR) accessory diamond crystal using an Invenio R FTIR spectrometer (Bruker). FTIR spectra were recorded within the wavenumber range from 4000 cm−1 to 400 cm−1 at 2 cm−1 resolution. In order to correctly subtract the background and, hence, obtain less noisy spectra, the solvent (0.9% NaCl water solution) was used at room temperature for the background measurement.
2.8. Solubility Assays
Thermodynamic solubility measurements were performed in an Invenio R FTIR spectrometer (Bruker) after equilibrating the solids in a 0.9% NaCl water solution [
8] under stirring at 500 rpm for 24 h. After the equilibrating time, the suspensions were filtrated through a 20 µm filter, and the resulting clear solution was analysed by FTIR. Then, the solid was analysed by PXRD to study the phase stability after equilibrium, resulting for (lidhcl)
2(res) and (lidhcl)
2(hq) that the cocrystal form remained stable after 24 h but not for (lidhcl)
2(pyr). A calibration curve was built with different (lidhcl) concentrations [
18,
19] obtaining linear models with
R2 values greater than 0.99, which were used to calculate the thermodynamic solubility. The peak used for the calibration curves was the area between 1712 cm
−1 and 1612 cm
−1, corresponding to (lidhcl), where there was no interference of any coformer.
3. Results and Discussion
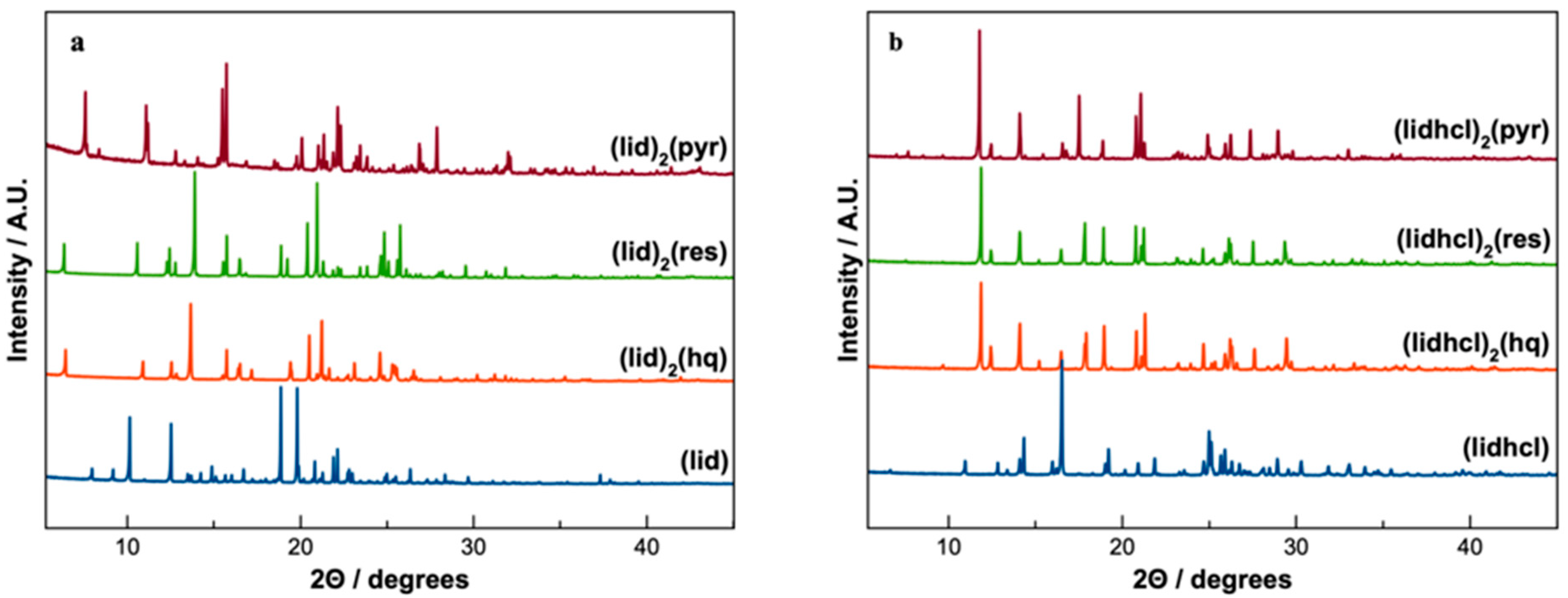
Six new phases were obtained for (lid) and (lidhcl) with hydroquinone, resorcinol, and pyrogallol. Only the phases with (lidhcl) demonstrated improved stability; accordingly, the physical characterisation is focused on these three new phases, after which the six phases are characterised structurally in order to study this stability differences. In all the cases, the correspondence between the SCXRD solved structures and the bulk powder was confirmed by PXRD (see
Supplementary Figure S1).
3.1. Stability Studies
After preparing slurries of the three new (lidhcl) phases, it can be clearly seen (
Figure S2) that (lidhcl)
2(res) and (lidhcl)
2(hq) were stable after 24 h stirring in water but not (lidhcl)
2(pyr), which transformed into a new phase that could not be structurally characterised but only identified as new form. Moreover, it was observed that crystal forms with (lid), compared to (lidhcl), drastically changed their colour (
Figure S2), which is a clear indicative of their poor stability with respect to oxidation.
Supplementary experiments were performed to evaluate the stability of (lidhcl) forms, in ageing conditions (40 °C and 75% relative humidity), where it can be observed that all three (lidhcl) new forms were stable after 2 months in ageing conditions (
Figure 2), which did not occur for (lid) forms, as can be easily seen from
Figure S2.
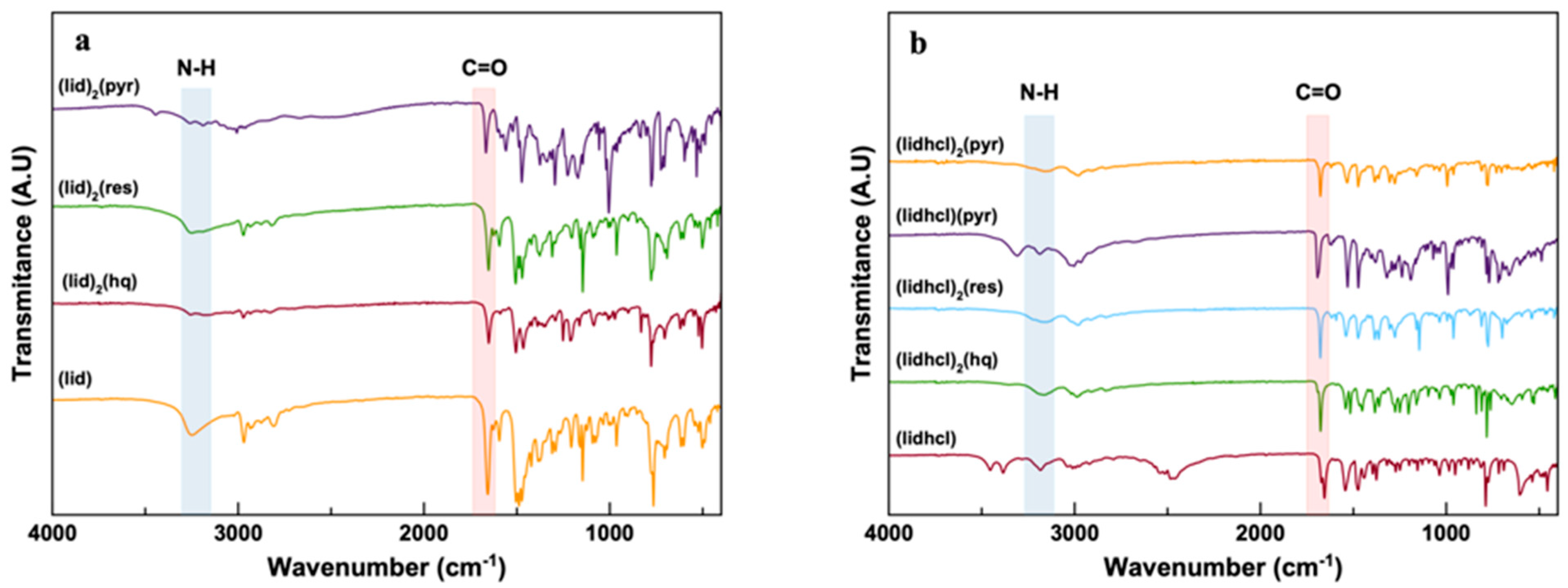
3.2. FTIR Spectroscopy and Solubility Measurements
Figure 3 shows the FTIR spectra for all six new phases obtained in this investigation. FTIR provides a fingerprint sign of each compound and wealthy information about the noncovalent interactions between acceptor and donor groups. Peak shifts can be found in the bands of the functional groups involved in the hydrogen bonds, namely, the carbonyl –C=O functional group of the amine group of the lidocaine and lidocaine hydrochloride molecule and –OH groups in the polyphenol molecules. The shifts in lidocaine’s –C=O group stretching vibration occurred from 1661 cm
−1 to 1623–1620 cm
−1 for the new crystalline forms obtained, and from 1680 cm
−1 to 1677–1670 cm
−1 for lidocaine hydrochloride. These shifts demonstrate the hydrogen bonding between the –C=O group of lidocaine molecules and the –OH groups of the polyphenols [
7], which as further confirmed by the structure solution.
In order to measure the solubility of the new (lidhcl) forms, FTIR measurements were performed to avoid the overlapping observed in the UV spectra, due to the ability of FTIR to show us isolated peaks from each component, allowing us to construct a calibration curve with the intensity of an unambiguous peak of (lidhcl).
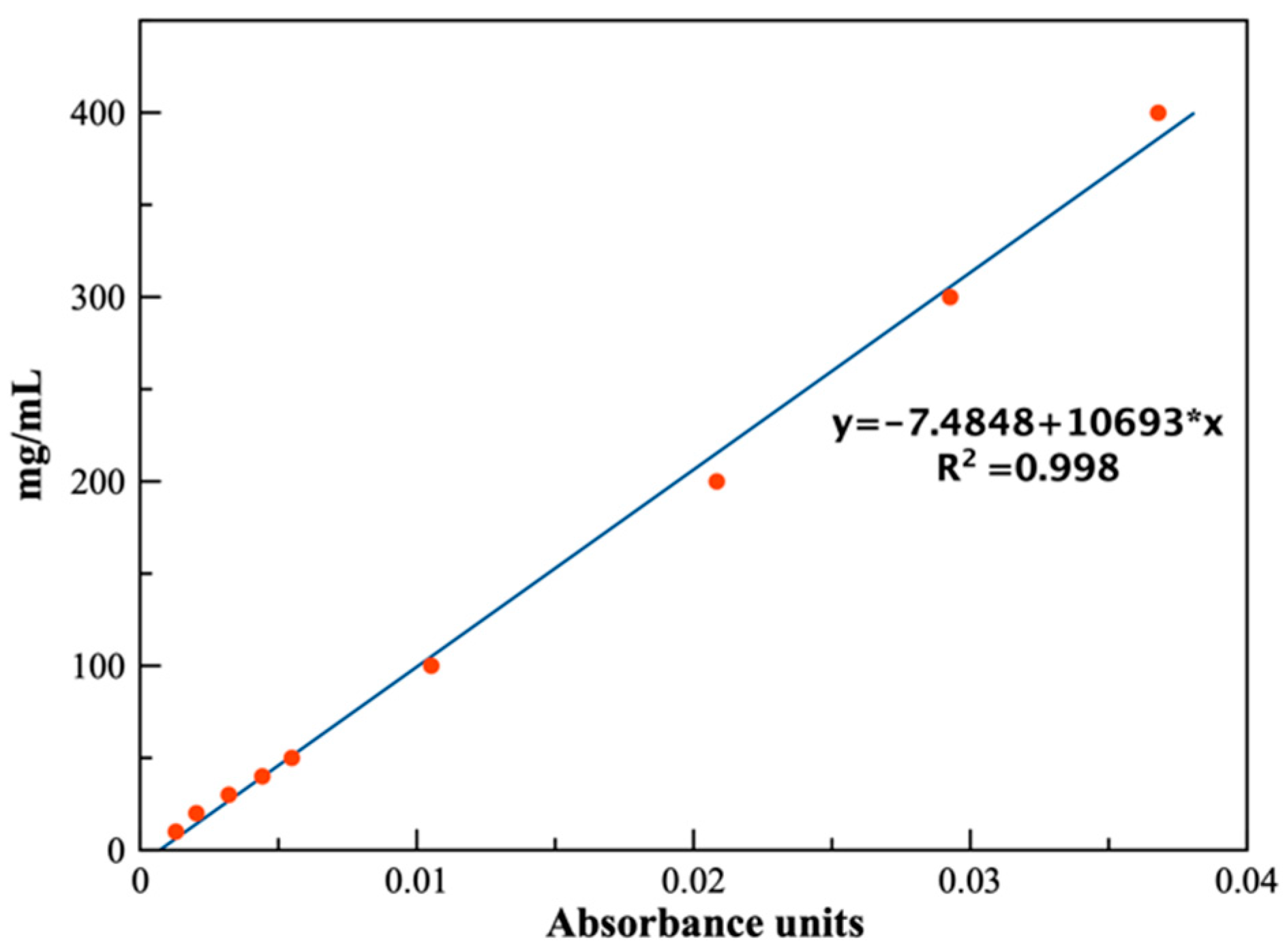
A calibration curve was built with different (lidhcl) concentrations [
18,
19] obtaining linear models with
R2 values greater than 0.99 (
Figure 4), which were used to calculate the apparent solubility. The peak used for the calibration curves was the area between 1712 cm
−1 and 1612 cm
−1 corresponding to (lidhcl), where there was no interference of any of the used coformers.
The constructed calibration curve is shown in
Figure 4, where the model’s excellent agreement with the experimental concentrations can be appreciated. Each concentration was prepared and measured in duplicate to assure reproducibility, and the mean value is represented.
Table 2 presents the apparent solubilities calculated from FTIR data for each new (lidhcl) phase.
The solubility decreased in the new phases with respect to reported values for
(lidhcl) [
9], generating
(lidhcl) forms with an interesting path to modulate solubility.
3.3. X-ray Diffraction
Powder X-ray diffraction patterns are a distinctive fingerprint of the crystalline phases. It can be confirmed from
Figure 5 how the PXRD patterns of the new
(lid) and
(lidhcl) phases were entirely different from the starting API, ensuring the appearance of new crystalline phases. Additionally, the comparison of the simulated PXRD data from SCXRD solved structures with the observed patterns obtained for each bulk powder obtained from LAG experiments could confirm the new obtained phases (
Figure S1).
From the single crystals obtained for each new phase, structure solutions were achieved, and these structures were analysed to extract information that could relate stability to the structure.
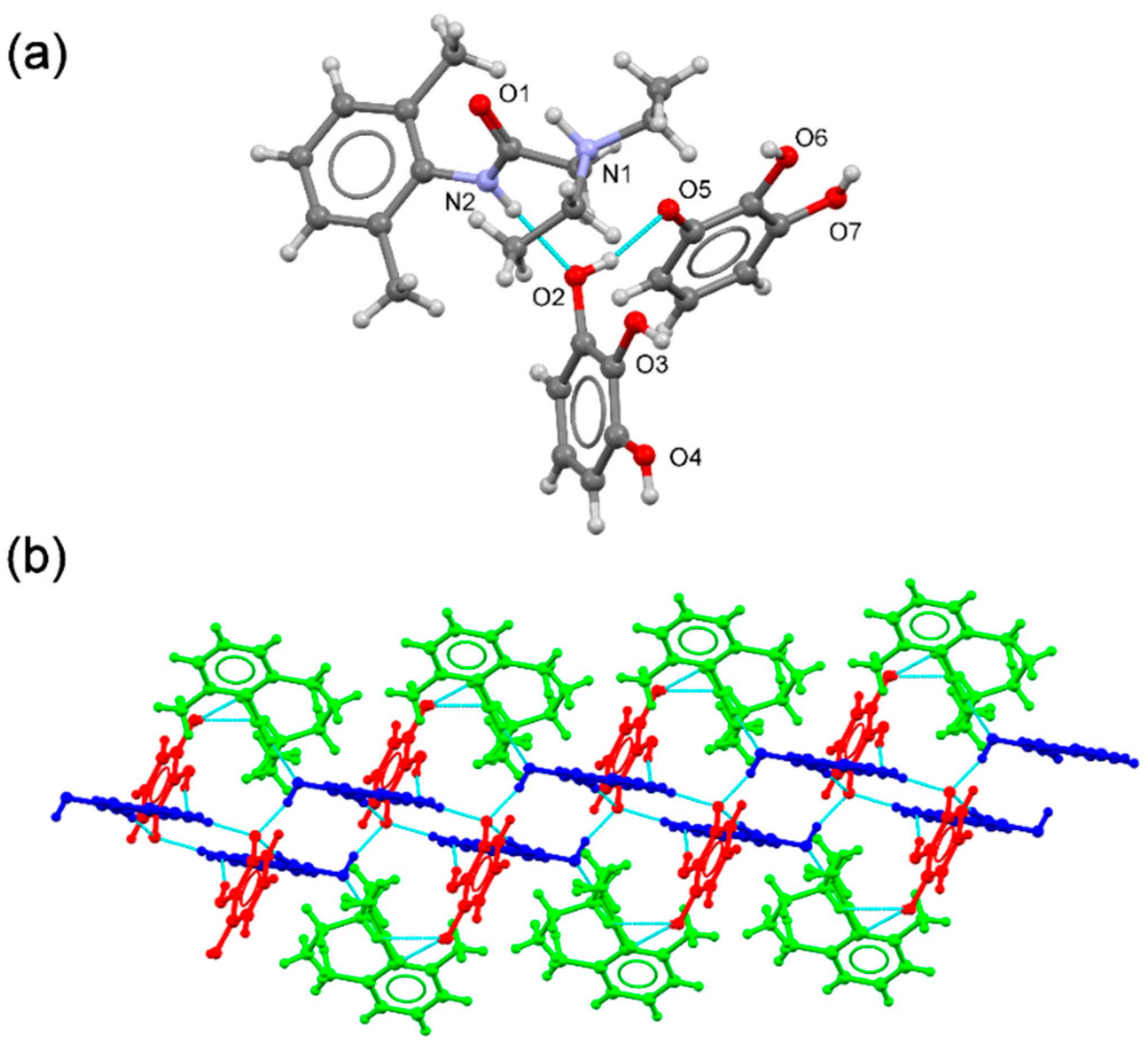
Cocrystals of lidocaine free base (lid) with two di-hydroxy isomers (hydroquinone, hq and resorcinol, res) and one tri-hydroxybenzene (pyrogallol, pyr) coformer were cocrystallised, and their crystal structure was determined. To evaluate the effect of the chloride ion on the stability and properties performance of lidocaine multicomponent solid forms, analogue cocrystals were also obtained using lidocaine hydrochloride salt (lidhcl). The structural features of both free base and ionic lidocaine systems are discussed in this section.
3.3.1. (lid/hq) Systems
The asymmetric unit of
(lid)2(hq) contains one
(lid) molecule and half an
(hq) molecule located at an inversion centre, leading to a 2:1 stoichiometric ratio (
Figure 6a). The hydroxy groups of
(hq) point in opposite directions, thus adopting a
trans conformation, forming discrete intermolecular OH(phenol)···O(carbonyl) hydrogen bonds with the
(2) graph set between the OH donor
(hq) and the carbonyl oxygen acceptor of the
(lid). These discrete units are further connected by centrosymmetric –C–H···π (2.968 Å) and –N–H···π (3.254 Å) interactions between
(lid) molecules, generating a 1D chain (
Figure 6b). Hydrophobic interactions involving methyl groups associate chains to build up the 3D structure.
The asymmetric unit of
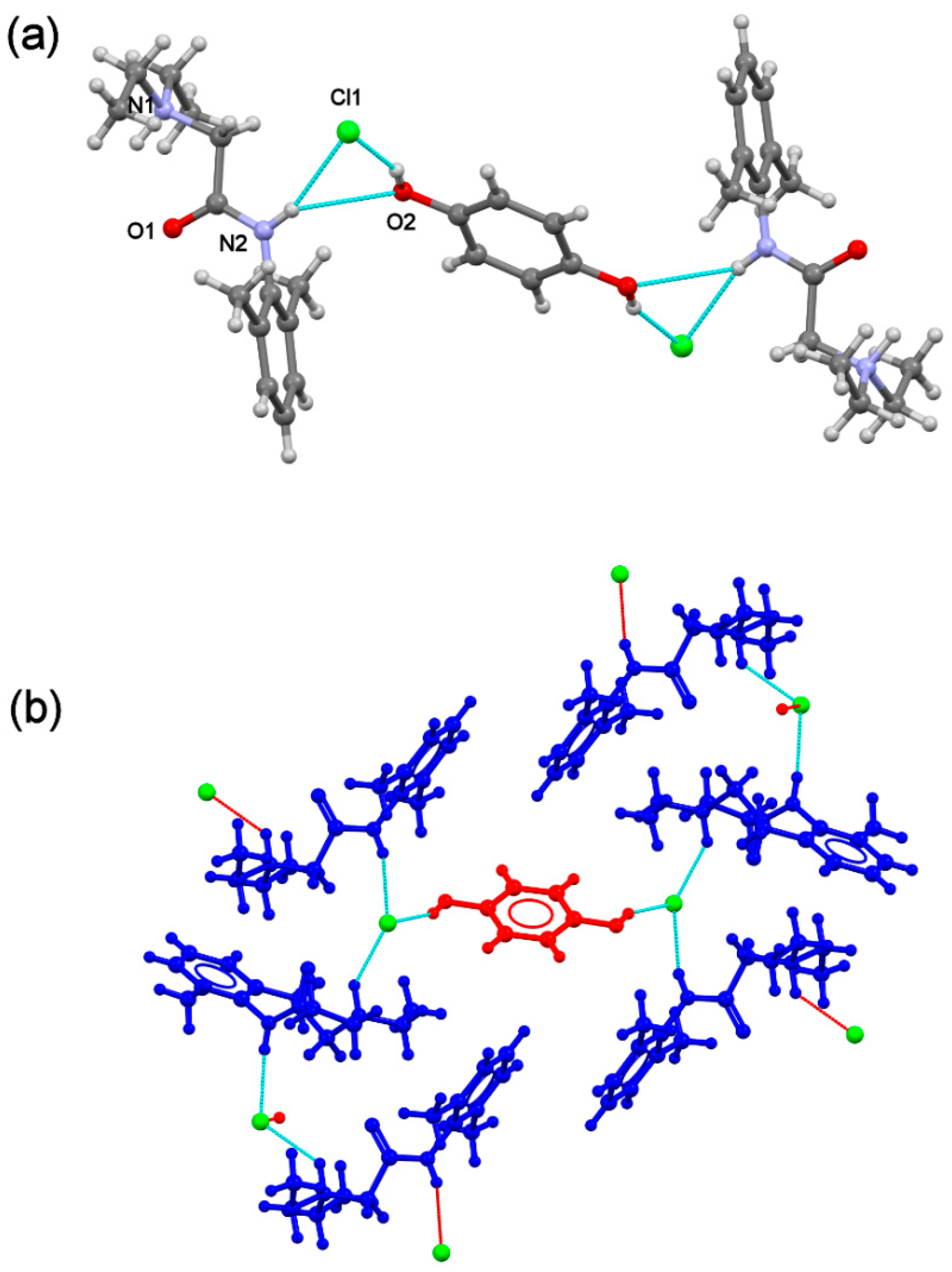
(lidhcl)2(hq) contains one
(lidhcl) salt and half an
(hq) molecule located at an inversion centre, with a 2:2:1 stoichiometric ratio considering all the components (
Figure 7a). Protonation of the N1 amine group results in a change in the
(lid) conformation, locating the amine groups in
trans conformation. The N2 amine group and the ammonium N1 group participate in electrostatic hydrogen bonds with the chloride ion, generating a chain reinforced by C–H···O hydrogen bonds. The
(hq) molecules connect chains via additional hydrogen bonds involving chloride ions, generating a 3D structure. The participation of chloride ions through noncovalent interactions results in a more compact crystal structure than the free base cocrystal analogue where each
(hq) molecule is surrounded by six
(lid+) cations, protecting the OH groups of
(hq) from oxidation (
Figure 7b).
3.3.2. (lid/res) Systems
(lid)2(res) crystallises in the triclinic P-1 space group. As in the
(lid)2(hq) cocrystal, the coformer is sitting on an inversion centre; however, in this compound, the
(res) coformer molecular symmetry does not exhibit inversion. Therefore, a disorder of the molecule about this particular special position is observed. The
(res) adopts a syn–syn conformation, pointing the –OH groups through the –C=O group of
(lid) (
(2) graph set) and establishing a 2:1 stoichiometric ratio (
Figure 8). These discrete units are connected in a similar way than in
(lid)2(hq) through shorter –C–H···π (2.864 Å) and –N–H···π (3.216 Å) interactions between
(lid) molecules, generating chains. Again, hydrophobic interactions involving methyl groups associate chains to build up the 3D structure. Crystal structure similarities in the
(lid) cocrystals [
20] with
(hq) and
(res) coformers could explain the template effect of
(lid)2(hq) solid when used as hetero-seeds during the synthesis of crystals of
(lid)2(res) (see
Section 2.3). The results obtained from crystal packing similarity calculations of one of the alternative positions of the disordered
(lid)2(res) structure and
(lid)2(hq) structure using Mercury [
21] showed that 14 out of 20 molecules were matched in the pairs of 2:1 cocrystals with a PXRD similarity index of 0.96745 and RMSD of 0.268 (
Figure 9). These results suggest that these pairs possess identical intermolecular interactions and lead to the same crystal packing [
22].
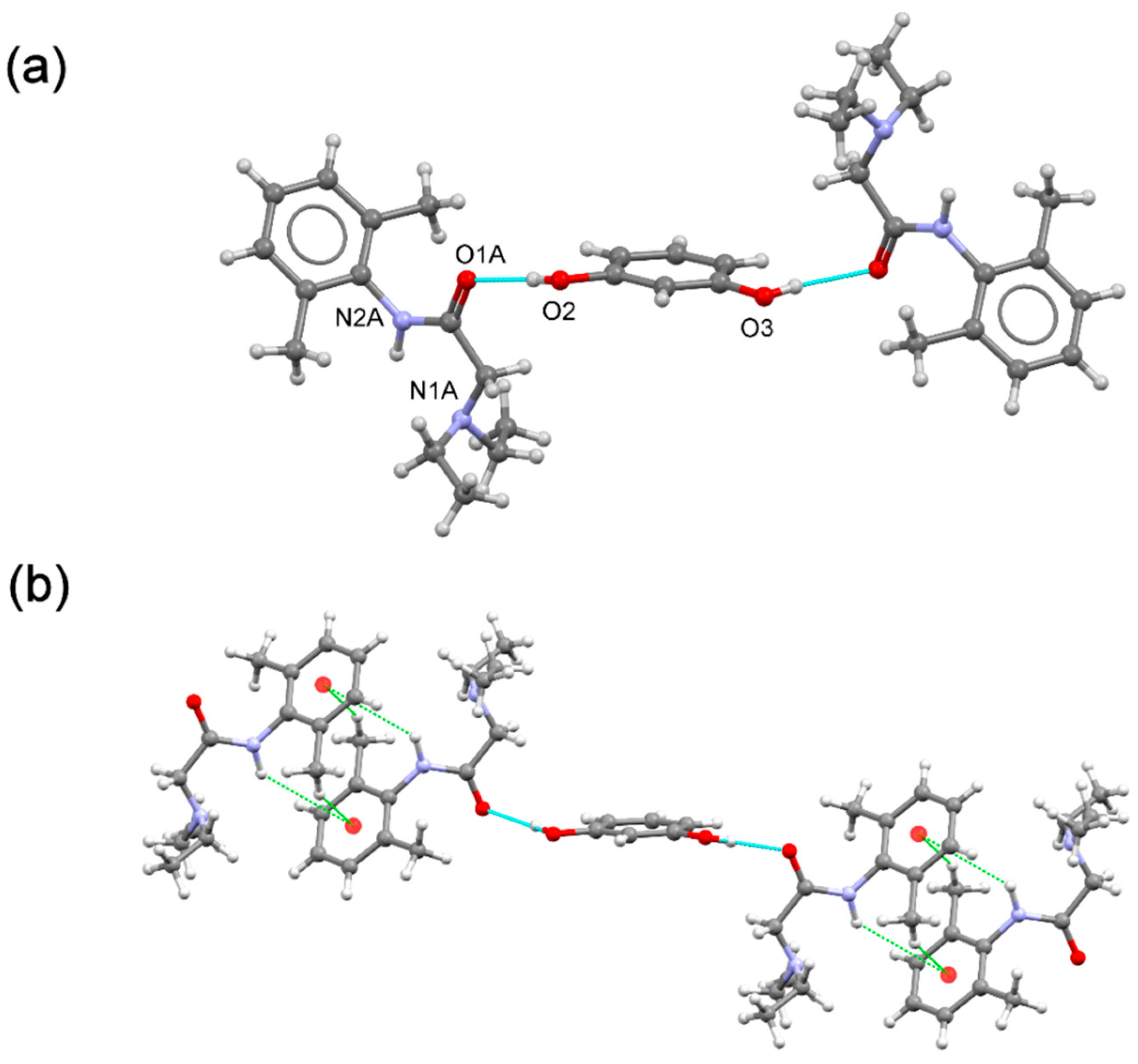
The
(lidhcl)2(res) ionic cocrystal [
20] contains one
(lidhcl) salt and half a
(res) molecule located in an inversion centre, giving a 2:1:1 stoichiometric ratio (
Figure 10a). As observed in the ionic cocrystal of
(lidhcl) with
(hq), protonation of
(lid) imposes a conformational change resulting in a chain structure build-up by electrostatic hydrogen bonds involving protonated lidocaine donor and acceptor groups, as well as chloride ions. Disordered
(res) molecules connect chains to generate the supramolecular 3D crystal structure. Similarly, as in the ionic cocrystal
(lidhcl)2(hq),
(res) coformer molecules are protected with
(lid+) cations, preventing them from oxidation (
Figure 10b).
3.3.3. (lid/pyr) Systems
The
(lid)2(pyr) is an ionic cocrystal and crystallises in the triclinic P-1 space group. Its asymmetric unit contains one
(lid+) molecule, one
(pyr-) anion, and one
(pyr) neutral molecule in a stoichiometric ratio 1:1:1 (
Figure 11a). Protonation of the tertiary amine in
(lid) imposes a change in its molecular conformation, pointing both amine protons in opposite directions with –N–C–C–N– torsion angles of −157.97° for
(lid)2(pyr) in comparison with −11.27° and 6.41° for
(lid)2(hq) and
(lid)2(res), respectively. The
(pyr) molecules adopt the anti-conformation [
22]. This conformation is energetically the least stable. However, considering the packing arrangement in
(lid)2(pyr), the anti-conformation ensures that each hydroxyl group of the pyrogallol moiety participates in hydrogen bonding interactions. In the crystal,
(pyr) molecules are connected to three
(pyr-) anions through H-bonding interactions generating chains running along the a axis. The
(lid+) molecules intercalate between two adjacent
(pyr−) anions through N–H···O(hydroxyl) and O–H···O(carbonyl) H-bonding and C–H interactions (
Figure 11b). In the crystal, C–H···π interactions connect these supramolecular chains to form the 3D structure.
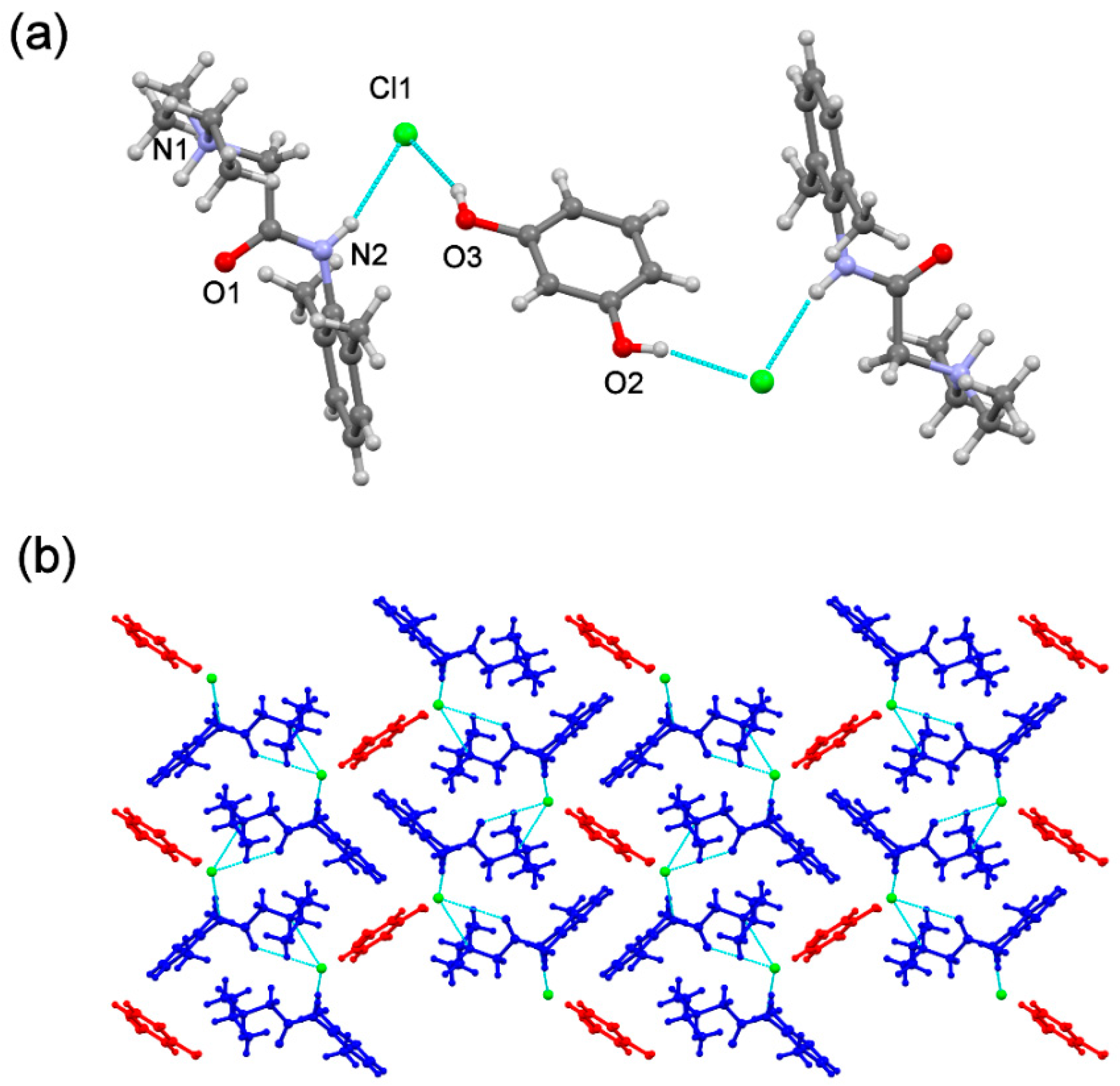
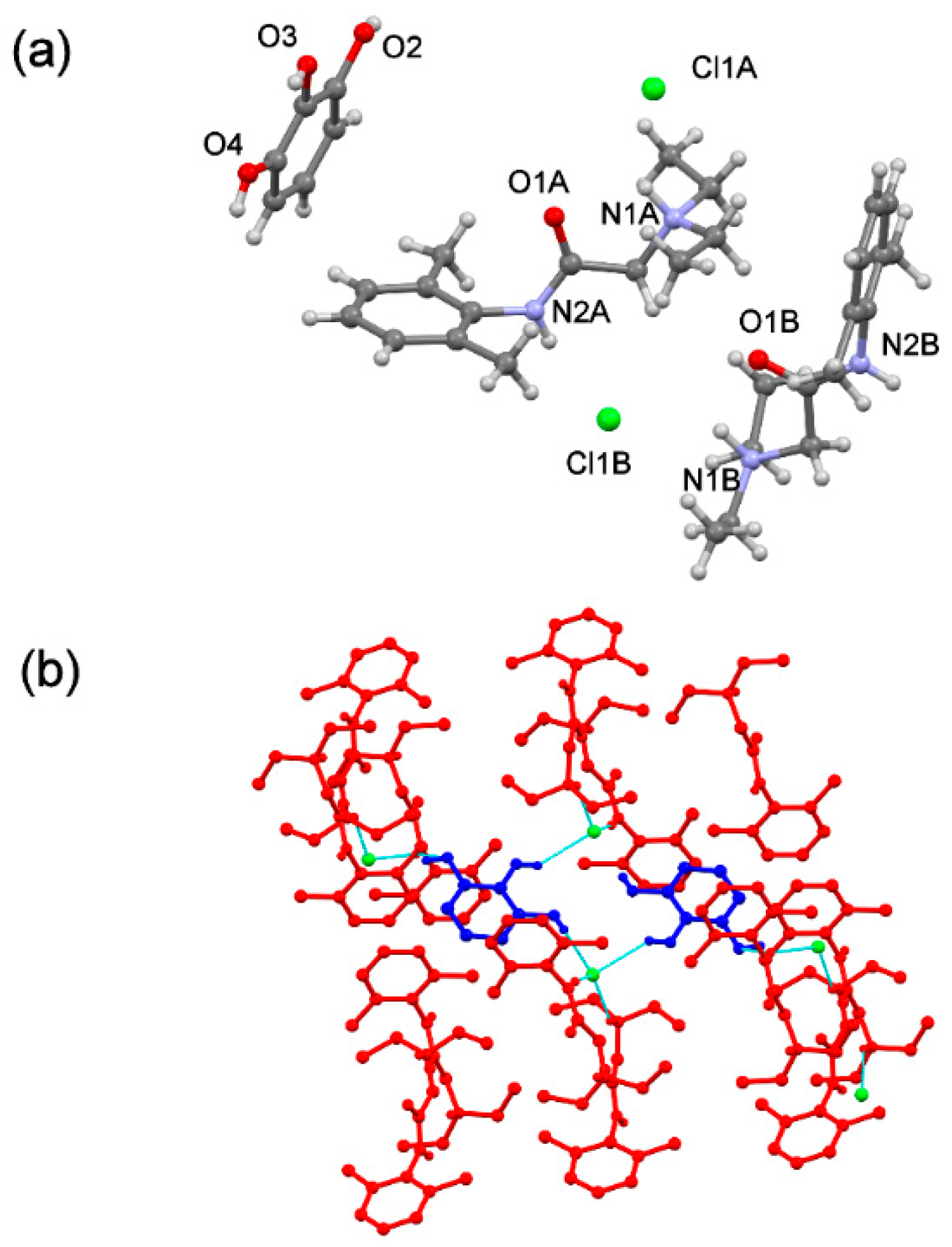
The
(lidhcl)2(pyr) is also an ionic cocrystal and crystallises in the monoclinic P21/c space group. Its asymmetric unit contains two
(lid+) two chloride ions and one
(pyr) molecule in a stoichiometric ratio 2:2:1 (
Figure 12a). As observed in the free base ionic cocrystal,
(pyr) adopts an anti-conformation where each hydroxyl group forms a hydrogen bond interaction with the corresponding chloride ion. In the crystal, Cl1A and Cl1B ions are H-bonded to four molecules, two
(pyr) and two
(lid+), and three molecules, one
(pyr) and two
(lid+) ions, respectively. This arrangement leads to efficient packing, shielding pairs of
(pyr) molecules that are surrounded with
(lid+) and Cl
− ions, thus protecting the coformer from oxidation (
Figure 12b).
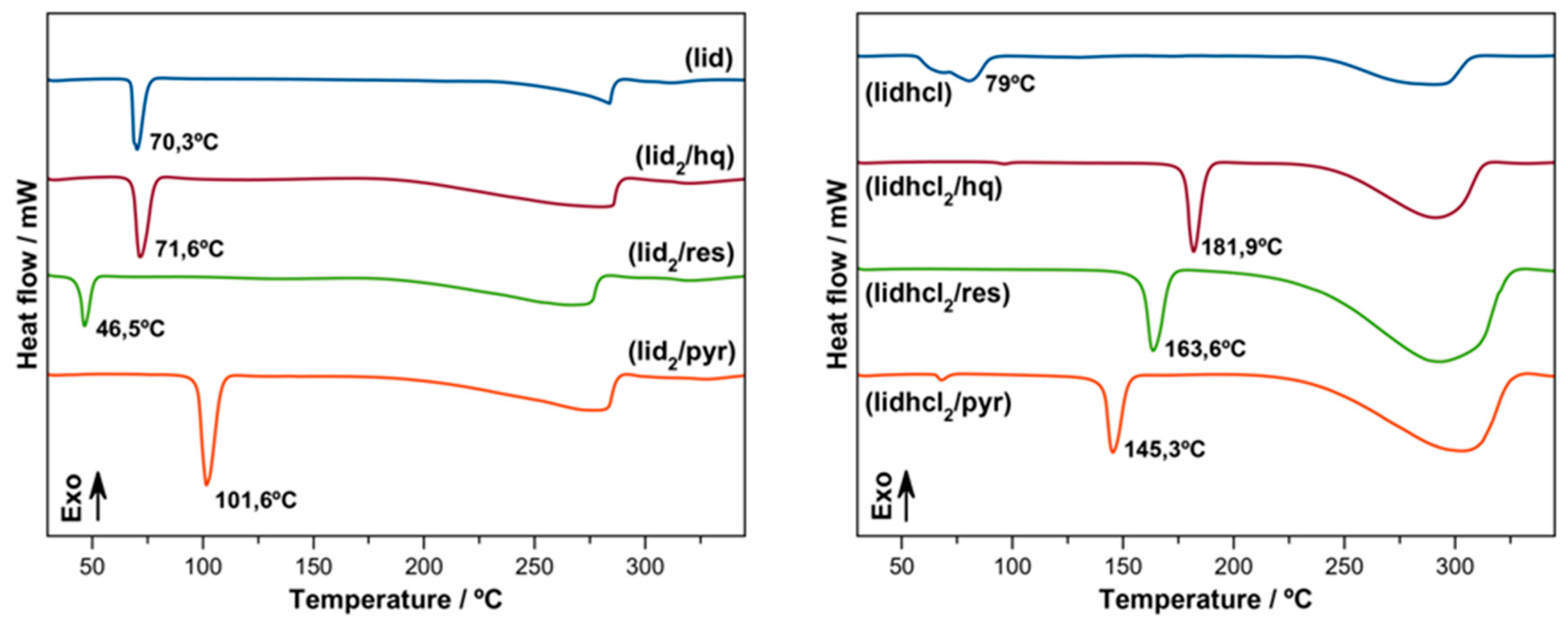
3.4. Thermal Analysis
DSC experiments were performed to evaluate the stability of the new phases, as well as to determine the melting points of the new multicomponent phases.
Figure 13 shows the DSC of the three
(lidhcl) and
(lid) phases.
In the case of the (lidhcl) phases, a decrease in the melting point can clearly be seen in the order (hq) > (res) > (pyr), with apparently no relation with structure effects.
There was a certain tendency with the measured solubilities, which also decreased in the same order, relating the melting point with the apparent solubility.
4. Conclusions
In this investigation, six new multicomponent forms, three for lidocaine and three for lidocaine hydrochloride, with hydroquinone, resorcinol, and pyrogallol, were obtained and characterised by different techniques. LAG allowed the synthesis of six new crystal forms, and the stability assays revealed the high stability of the (lidhcl) forms compared with (lid) phases, which were selected for further analysis. The SC-XRD structure solution allowed a comparative structural analysis, highlighting how (lidhcl) increases the stability of the new forms compared to (lid) through a steric protection effect. The structural information obtained revealed the important role that chloride ions play in the stabilisation of the (lidhcl) new multicomponent forms, allowing an improved oxidation behaviour.
There was a tendency between melting points and solubility for the new (lidhcl) phases. The solubility of the new crystal forms of (lidhcl) was lower than (lidhcl) itself, which, far from being a drawback, can be an interesting way to modulate the (lidhcl) solubility and stability, as can be concluded from the DSC of the new phases showing an increase in thermal stability. In this concern, the synthesis of molecular or ionic cocrystals can be taken as a valuable approach to tuning the properties of the API to adapt it to the requirements.


 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}