
Identification of Novel AXL Kinase Inhibitors Using Ligand-Based Pharmacophore Screening and Molecular Dynamics Simulations

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Methodology
2.1. Pharmacophore Modeling Based on Structure
2.2. Virtual Screening
2.3. Molecular Dynamics (MD) Simulations and MMPBSA Free Energies
3. Results and Discussion
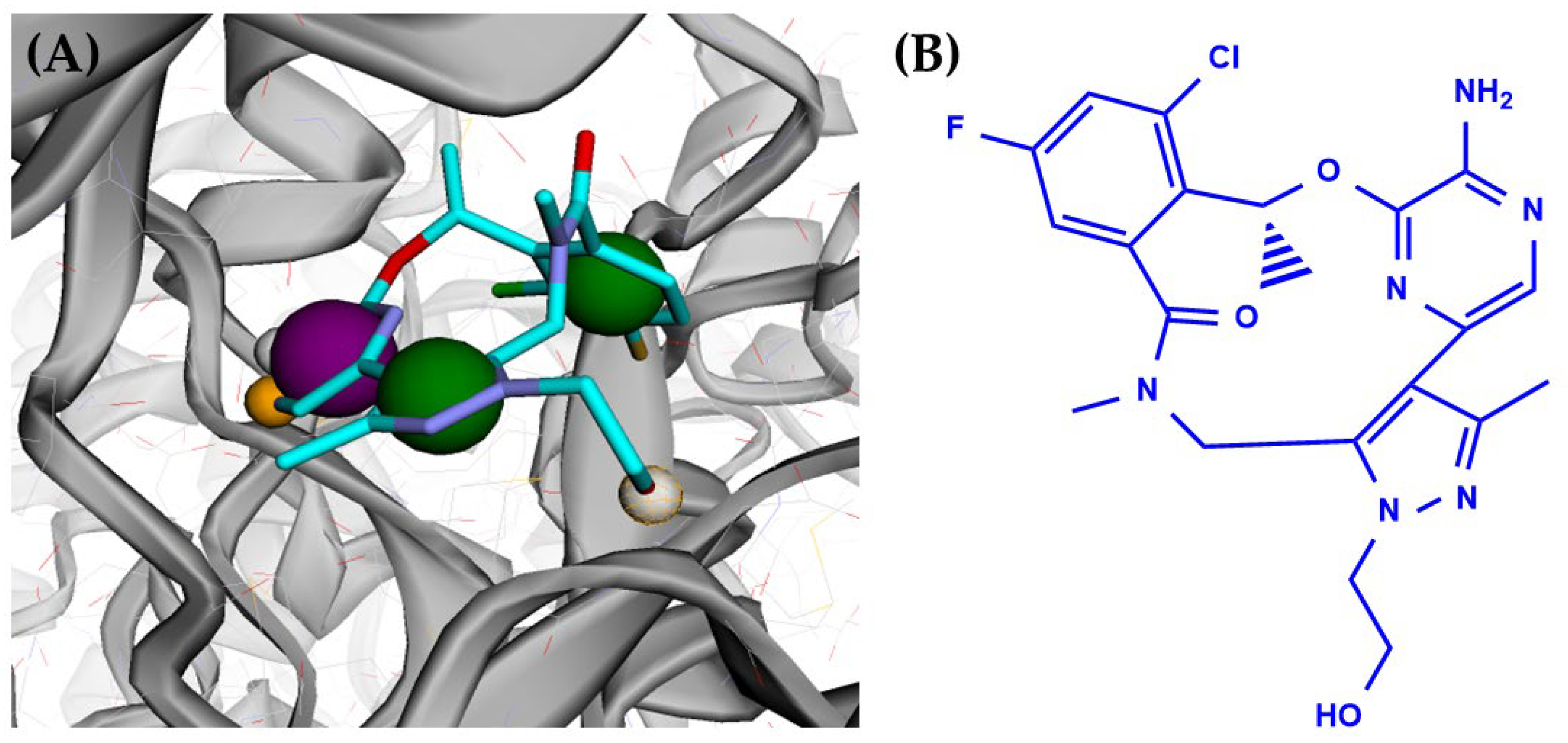
3.1. AXL Kinase Active Site Pharmacophore Modeling
3.2. Virtual Screening Analysis
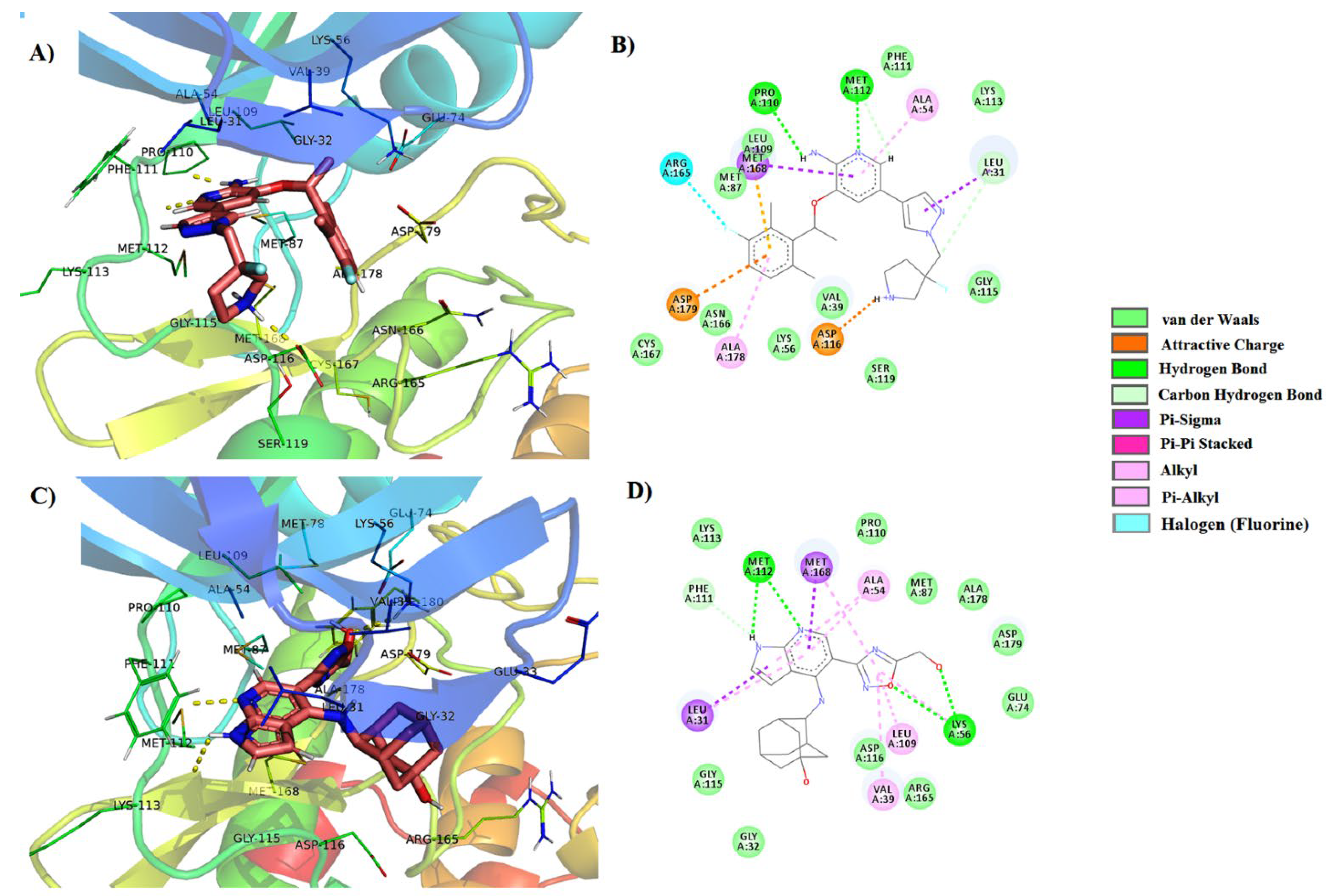
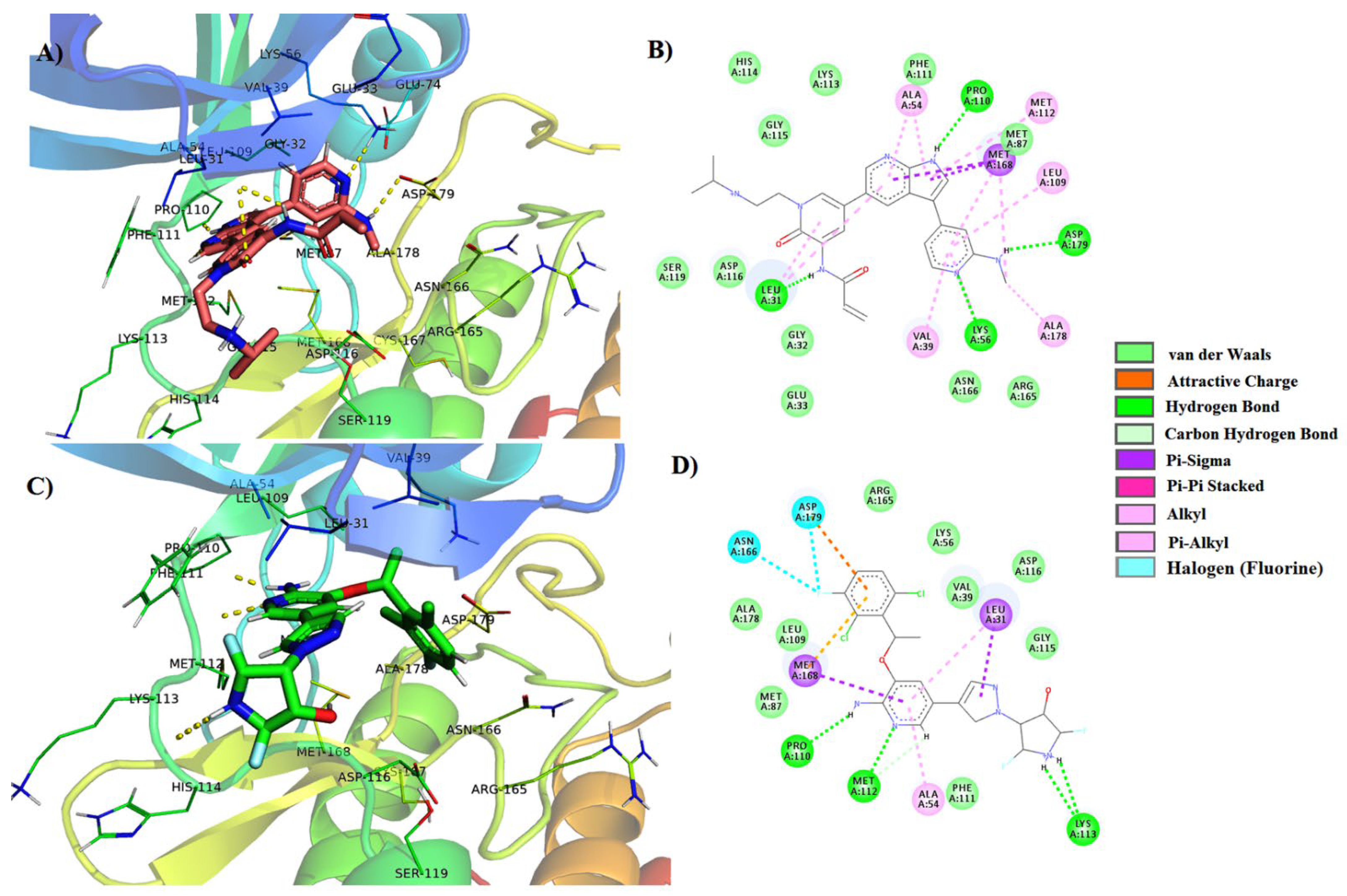
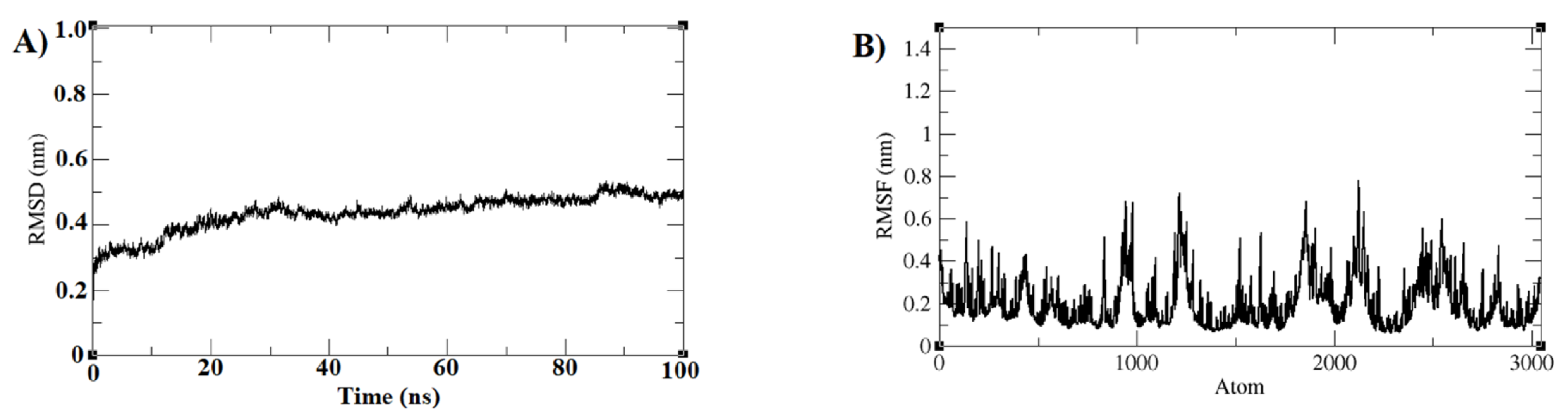
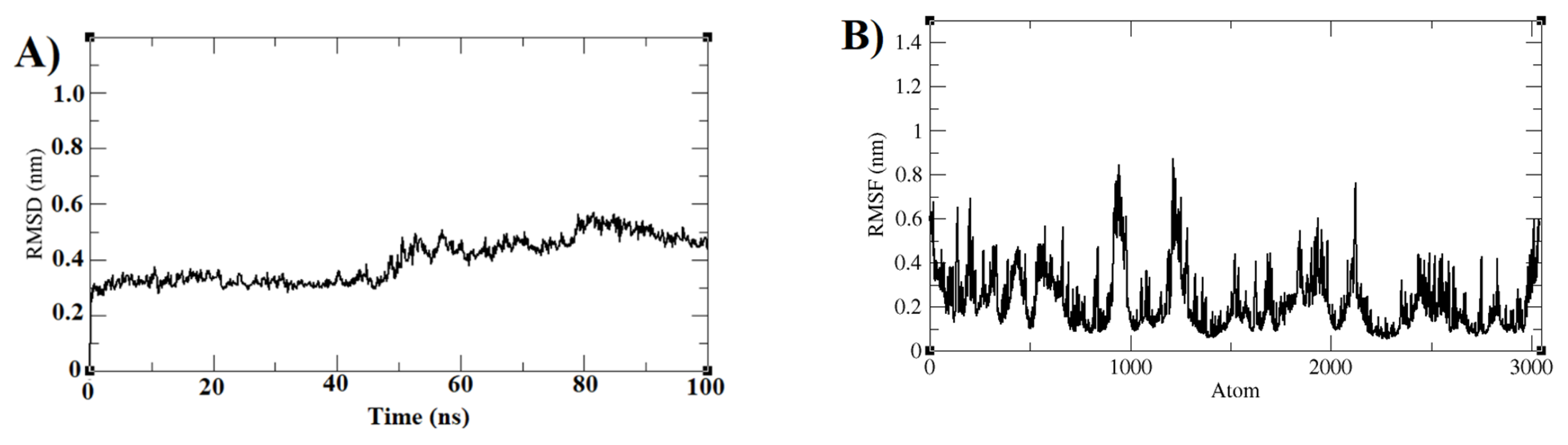
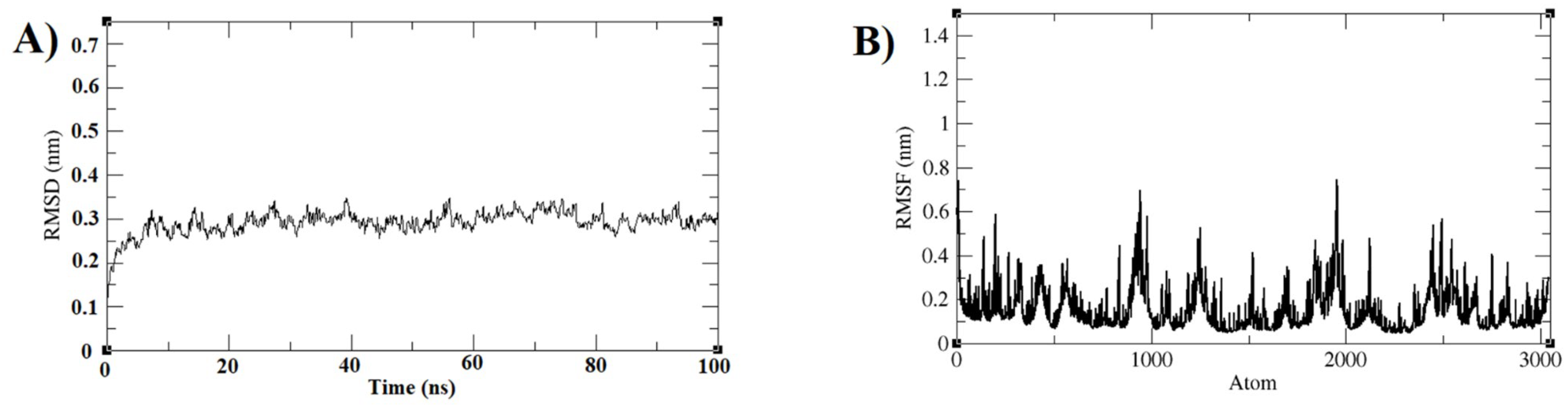
3.3. Molecular Dynamics Simulations
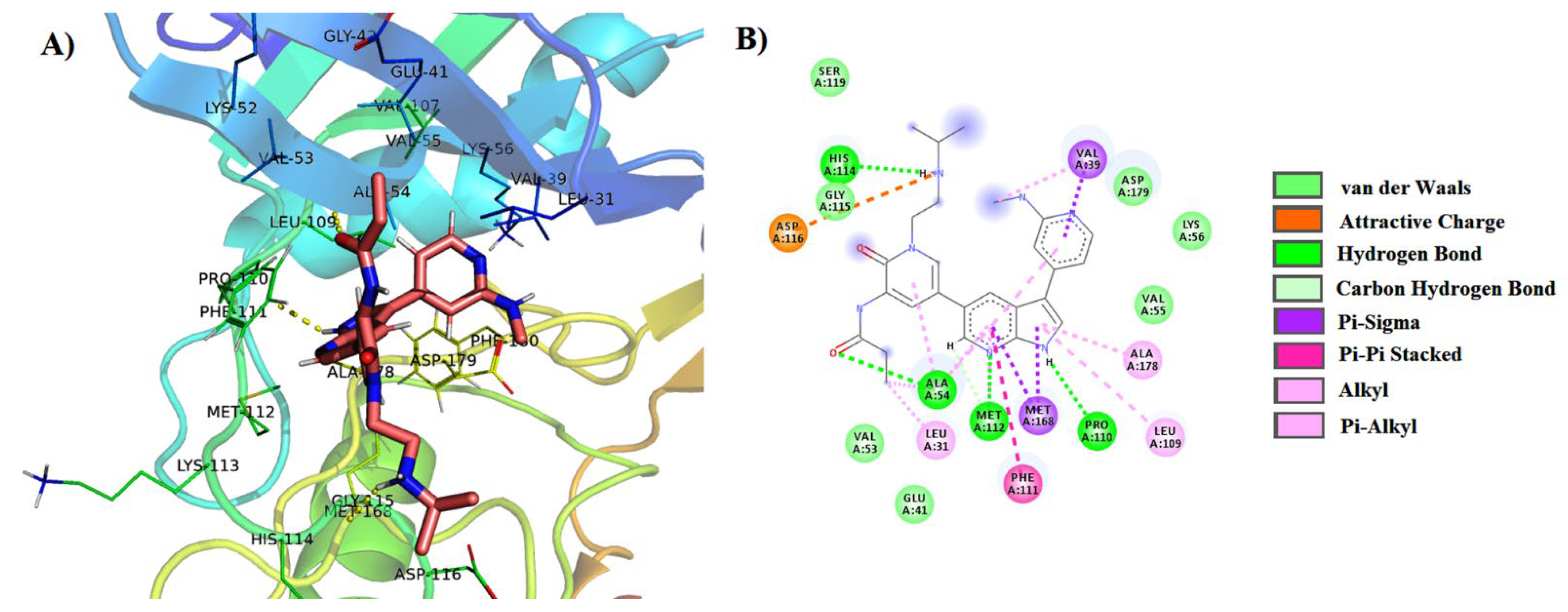
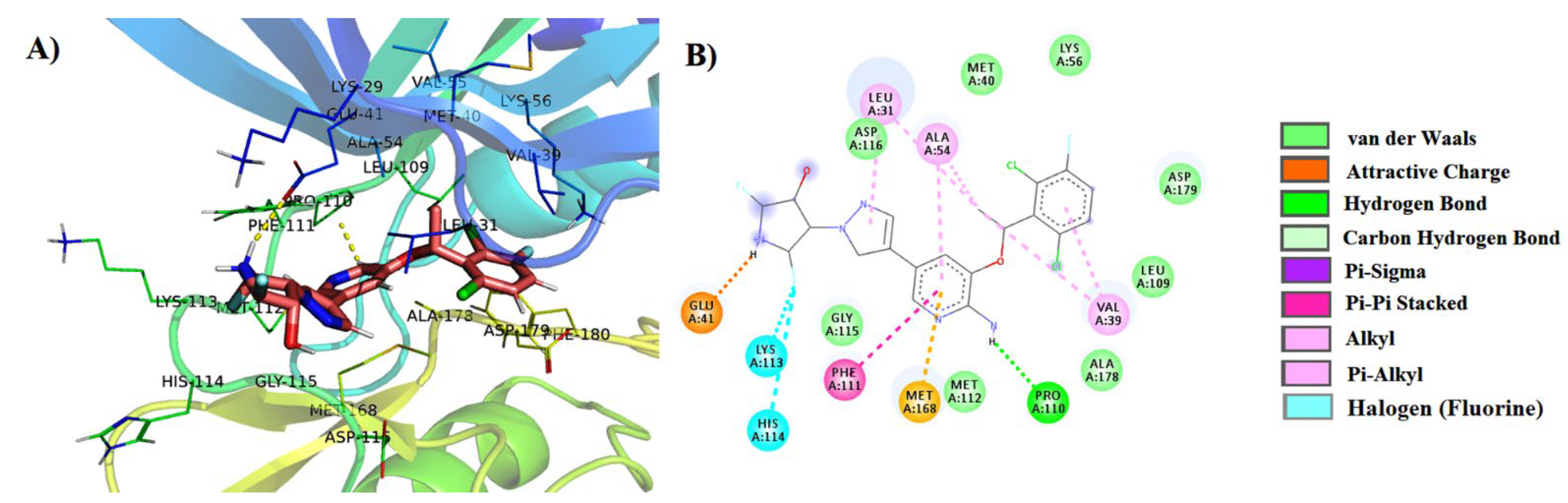
3.4. MMPBSA (Molecular Mechanics Poisson–Boltzmann Surface Area) Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arends, J. Metabolism in cancer patients. Anticancer Res. 2010, 30, 1863–1868. [Google Scholar] [PubMed]
- Mollard, A.; Warner, S.L.; Call, L.T.; Wade, M.L.; Bearss, J.J.; Verma, A.; Sharma, S.; Vankayalapati, H.; Bearss, D.J. Design, Synthesis, and Biological Evaluation of a Series of Novel AXL Kinase Inhibitors. ACS Med. Chem. Lett. 2011, 2, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Niu, Y.; Li, S.; Zu, X.; Xiao, M.; Yin, L.; Feng, J.; He, J.; Shen, Y. Identification of an AXL kinase inhibitor in triple-negative breast cancer by structure-based virtual screening and bioactivity test. Chem. Biol. Drug Des. 2022, 99, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Vouri, M.; Hafizi, S. TAM receptor tyrosine kinases in cancer drug resistance. Cancer Res. 2017, 77, 2775–2778. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Hjelle, B.; Bishop, J.M. Transforming genes in chronic myelogenous leukemia. Proc. Natl. Acad. Sci. USA 1988, 85, 1952–1956. [Google Scholar] [CrossRef]
- O’Bryan, J.P.; Frye, R.; Cogswell, P.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., III; Le Beau, M.; Earp, H.; Liu, E. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar]
- Neubauer, A.; O’BRYAN, J.; Fiebeler, A.; Schmidt, C.; Huhn, D.; Liu, E. Axl, a novel receptor tyrosine kinase isolated from chronic myelogenous leukemia. Semin. Hematol. 1993, 30, 3. [Google Scholar]
- Li, Y.; Ye, X.; Tan, C.; Hongo, J.-A.; Zha, J.; Liu, J.; Kallop, D.; Ludlam, M.; Pei, L. Axl as a potential therapeutic target in cancer: Role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009, 28, 3442–3455. [Google Scholar] [CrossRef]
- Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007, 26, 3909–3919. [Google Scholar] [CrossRef]
- Gjerdrum, C.; Tiron, C.; Høiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.; Boström, A.-K.; Ljungberg, B.; Axelson, H.; Dahlbäck, B. Gas6 and the receptor tyrosine kinase Axl in clear cell renal cell carcinoma. PLoS ONE 2009, 4, e7575. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.; Martuszewska, D.; Johansson, M.; Ekman, C.; Hafizi, S.; Ljungberg, B.; Dahlback, B. Differential expression of Axl and Gas6 in renal cell carcinoma reflecting tumor advancement and survival. Clin. Cancer Res. 2009, 15, 4742–4749. [Google Scholar] [CrossRef] [PubMed]
- Koorstra, J.-B.M.; Karikari, C.; Feldmann, G.; Bisht, S.; Leal-Rojas, P.; Offerhaus, G.J.A.; Alvarez, H.; Maitra, A. The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol. Ther. 2009, 8, 618–626. [Google Scholar] [CrossRef]
- Ueno, Y.; Kaneko, N.; Ueno, Y.; Tanaka, R.; Cho, K.; Saito, R.; Kondoh, Y.; Shimada, I.; Kuromitsu, S. ASP2215, a novel FLT3/AXL inhibitor: Preclinical evaluation in acute myeloid leukemia (AML). Am. Soc. Clin. Oncol. 2014, 32, 7070. [Google Scholar] [CrossRef]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef]
- Hart, C.D.; De Boer, R.H. Profile of cabozantinib and its potential in the treatment of advanced medullary thyroid cancer. OncoTargets Ther. 2013, 6, 1. [Google Scholar]
- You, W.-K.; Sennino, B.; Williamson, C.W.; Falcón, B.; Hashizume, H.; Yao, L.-C.; Aftab, D.T.; McDonald, D.M. VEGF and c-Met Blockade Amplify Angiogenesis Inhibition in Pancreatic Islet CancerAmplified Antiangiogenic Action in Tumors. Cancer Res. 2011, 71, 4758–4768. [Google Scholar] [CrossRef]
- Qian, F.; Engst, S.; Yamaguchi, K.; Yu, P.; Won, K.-A.; Mock, L.; Lou, T.; Tan, J.; Li, C.; Tam, D. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009, 69, 8009–8016. [Google Scholar] [CrossRef]
- Yan, S.B.; Peek, V.L.; Ajamie, R.; Buchanan, S.G.; Graff, J.R.; Heidler, S.A.; Hui, Y.-H.; Huss, K.L.; Konicek, B.W.; Manro, J.R. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Investig. New Drugs 2013, 31, 833–844. [Google Scholar] [CrossRef]
- Miknyoczki, S.; Cheng, M.; Hudkins, R.; Angeles, T.; Aimone, L.; Husten, J.; Qian, J.; Murthy, S.; Conners, T.; Bendesky, R. Abstract C275: CEP-40783: A potent and selective AXL/c-Met inhibitor for use in breast, non-small cell lung (NSCLC), and pancreatic cancers. Mol. Cancer Ther. 2013, 12, C275. [Google Scholar] [CrossRef]
- Wang, Y.; Xing, L.; Ji, Y.; Ye, J.; Dai, Y.; Gu, W.; Ai, J.; Song, Z. Discovery of a potent tyrosine kinase AXL inhibitor bearing the 3-((2, 3, 4, 5-tetrahydro-1H-benzo [d] azepin-7-yl) amino) pyrazine core. Bioorganic Med. Chem. Lett. 2019, 29, 836–838. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef] [PubMed]
- Pirhadi, S.; Shiri, F.; Ghasemi, J.B. Methods and applications of structure based pharmacophores in drug discovery. Curr. Top. Med. Chem. 2013, 13, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Shiri, F.; Pirhadi, S.; Rahmani, A. Identification of new potential HIV-1 reverse transcriptase inhibitors by QSAR modeling and structure-based virtual screening. J. Recept. Signal Transduct. 2018, 38, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Modeling 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Gaur, A.S.; Nagamani, S.; Tanneeru, K.; Druzhilovskiy, D.; Rudik, A.; Poroikov, V.; Sastry, G.N. Molecular property diagnostic suite for diabetes mellitus (MPDSDM): An integrated web portal for drug discovery and drug repurposing. J. Biomed. Inform. 2018, 85, 114–125. [Google Scholar] [CrossRef]
- Gajiwala, K.S.; Grodsky, N.; Bolaños, B.; Feng, J.; Ferre, R.; Timofeevski, S.; Xu, M.; Murray, B.W.; Johnson, T.W.; Stewart, A. The Axl kinase domain in complex with a macrocyclic inhibitor offers first structural insights into an active TAM receptor kinase. J. Biol. Chem. 2017, 292, 15705–15716. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Tanneeru, K.; Balla, A.R.; Guruprasad, L. In silico 3D structure modeling and inhibitor binding studies of human male germ cell-associated kinase. J. Biomol. Struct. Dyn. 2015, 33, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Tanneeru, K.; Guruprasad, L. Ponatinib is a pan-BCR-ABL kinase inhibitor: MD simulations and SIE study. PLoS ONE 2013, 8, e78556. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Yan, R.; Jiang, Y.; Li, Z.; Zhang, J.; Wu, X. Insight into binding mechanisms of EGFR allosteric inhibitors using molecular dynamics simulations and free energy calculations. J. Biomol. Struct. Dyn. 2019, 37, 4384–4394. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, S.; Habeeb, S. Combined pharmacophore, virtual screening and molecular dynamics studies to identify Bruton’s tyrosine kinase inhibitors. J. Biomol. Struct. Dyn. 2018, 36, 4320–4337. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; Van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Uppula, P. Exploration of Conformations, Analysis of Protein and Biological Significance of Histidine Dimers. ChemistrySelect 2018, 3, 3070–3078. [Google Scholar] [CrossRef]
- Purushotham, U.; Sastry, G.N. A comprehensive conformational analysis of tryptophan, its ionic and dimeric forms. J. Comput. Chem. 2014, 35, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, U.; Zipse, H.; Sastry, G.N. A first-principles investigation of histidine and its ionic counterparts. Theor. Chem. Acc. 2016, 135, 1–16. [Google Scholar] [CrossRef]
- Purushotham, U.; Vijay, D.; Narahari Sastry, G. A computational investigation and the conformational analysis of dimers, anions, cations, and zwitterions of L-phenylalanine. J. Comput. Chem. 2012, 33, 44–59. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | PubChem ID | Molecule Structure | Binding Score |
|---|---|---|---|
| 1 | PubChem-78160848 |  | −8.3 |
| 2 | PubChem-122421875 |  | −7.5 |
| 3 | PubChem-76973938 |  | −8.2 |
| 4 | PubChem-57928218 |  | −7.9 |
| 5 | PubChem-54673081 |  | −8.3 |
| 6 | PubChem-126726905 |  | −7.9 |
| 7 | PubChem-68651602 |  | −7.9 |
| 8 | PubChem-76167946 |  | −7.8 |
| 9 | PubChem-25224351 |  | −7.8 |
| 10 | PubChem-68650375 |  | −7.7 |
| S.No | Molecule | Bonds between Protein and Ligand | Type of Bond | Distance in Å |
|---|---|---|---|---|
| 1 | PubChem-76973938 | LIG:NH–Pro110 LIG:N–Met112 LIG–Met168 LIG–Leu31 LIG–Asp179 LIG–Met168 LIG–Arg165 | Hydrogen Bond Hydrogen Bond Pi-Sigma Pi-Sigma Pi-Anion Pi-Sulfur Halogen | 2.09 2.18 3.44 3.77 4.71 5.87 6.72 |
| 2 | PubChem-57928218 | LIG: NH–Met112 LIG:N–Met112 LIG:OH–Lys56 LIG:O–Lys56 LIG–Leu31 LIG–Met168 LIG–Ala54 LIG–Leu109 | Hydrogen Bond Hydrogen Bond Hydrogen Bond Hydrogen Bond Pi-Sigma Pi-Sigma Pi-Alkyl Pi-Alkyl | 2.69 2.41 2.49 2.87 4.00 3.48 4.02 4.93 |
| 3 | PubChem-122421875 | LIG:N–Lys56 LIG:NH–Pro110 LIG:NH–Asp179 LIG:NH–Leu31 LIG–Met168 LIG–Ala54 LIG–Val39 LIG–Leu109 LIG- Met112 | Hydrogen Bond Hydrogen Bond Hydrogen Bond Hydrogen Bond Pi-Sigma Pi-Alkyl Pi-Alkyl Pi-Alkyl Pi-Alkyl | 2.51 2.05 2.38 2.29 3.73 4.73 4.99 4.28 5.49 |
| 4 | PubChem-78160848 | LIG:NH–Pro110 LIG:NH–Lys113 LIG:N–Met112 LIG–Met168 LIG–Leu31 LIG–Asp179 LIG–Met168 LIG–Asn166 LIG–Asp179 | Hydrogen Bond Hydrogen Bond Hydrogen Bond Pi-Sigma Pi-Sigma Pi-Anion Pi-Sulfur Fluorine Fluorine | 2.08 2.60 2.24 3.45 5.45 4.55 5.47 4.23 3.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagamalla, L.; Kumar, J.V.S.; Shaik, M.R.; Sanjay, C.; Alsamhan, A.M.; Kasim, M.A.; Alwarthan, A. Identification of Novel AXL Kinase Inhibitors Using Ligand-Based Pharmacophore Screening and Molecular Dynamics Simulations. Crystals 2022, 12, 1158. https://doi.org/10.3390/cryst12081158
Nagamalla L, Kumar JVS, Shaik MR, Sanjay C, Alsamhan AM, Kasim MA, Alwarthan A. Identification of Novel AXL Kinase Inhibitors Using Ligand-Based Pharmacophore Screening and Molecular Dynamics Simulations. Crystals. 2022; 12(8):1158. https://doi.org/10.3390/cryst12081158
Chicago/Turabian StyleNagamalla, Lavanya, J. V. Shanmukha Kumar, Mohammed Rafi Shaik, Chintakindi Sanjay, Ali M. Alsamhan, Mohsin Ahmed Kasim, and Abdulrahman Alwarthan. 2022. "Identification of Novel AXL Kinase Inhibitors Using Ligand-Based Pharmacophore Screening and Molecular Dynamics Simulations" Crystals 12, no. 8: 1158. https://doi.org/10.3390/cryst12081158
APA StyleNagamalla, L., Kumar, J. V. S., Shaik, M. R., Sanjay, C., Alsamhan, A. M., Kasim, M. A., & Alwarthan, A. (2022). Identification of Novel AXL Kinase Inhibitors Using Ligand-Based Pharmacophore Screening and Molecular Dynamics Simulations. Crystals, 12(8), 1158. https://doi.org/10.3390/cryst12081158