
Crystal Structures of CuCl2·2H2O (Eriochalcite) and NiCl2∙6H2O (Nickelbischofite) at Low Temperature: Full Refinement of Hydrogen Atoms Using Non-Spherical Atomic Scattering Factors


Abstract
:1. Introduction
2. Results and Discussion
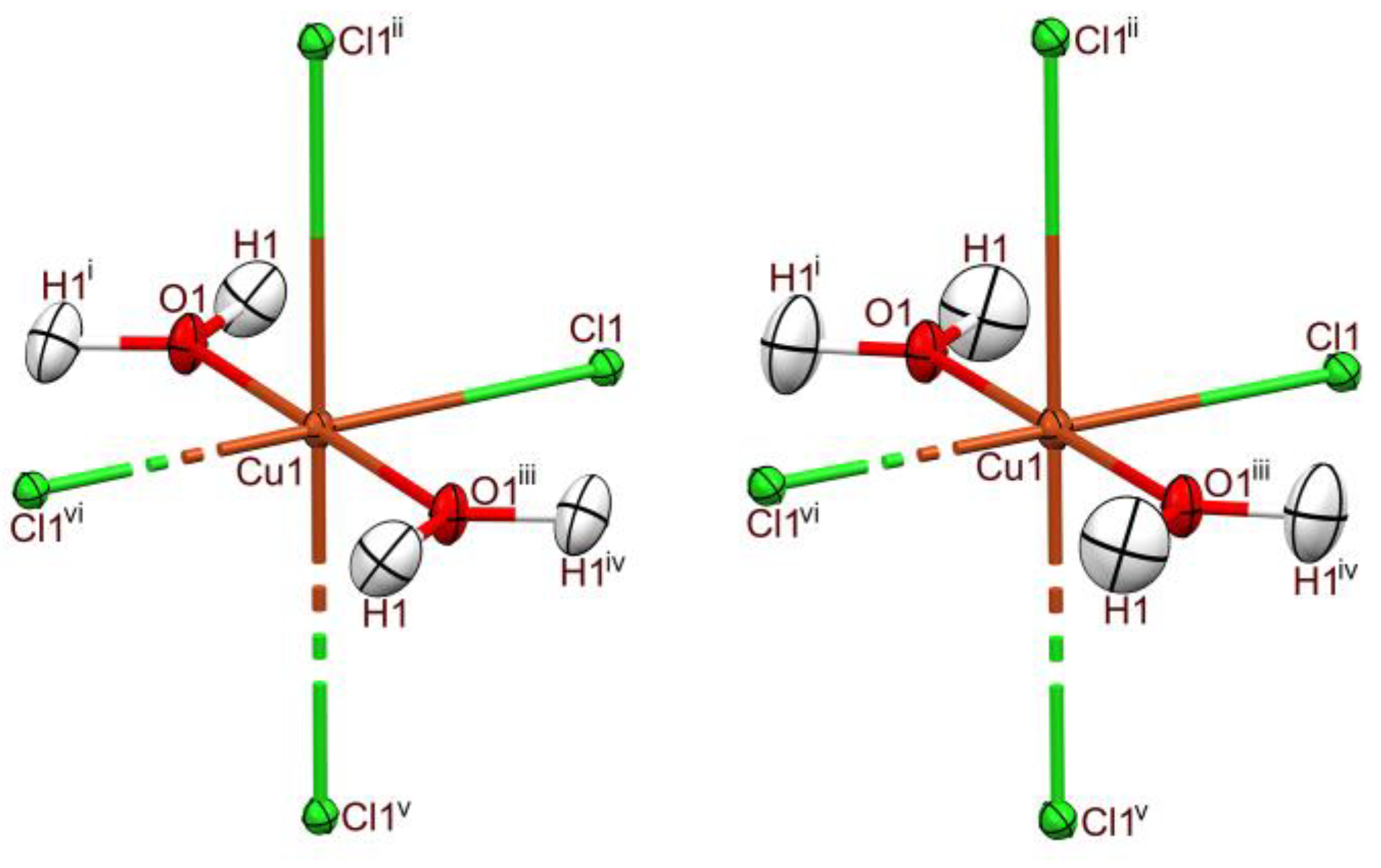
2.1. X-Ray Diffraction Structure of CuCl2∙2H2O
2.2. Comparison to Structure of Anhydrous CuCl2
2.3. X-ray Diffraction Structure of NiCl2∙6H2O
2.4. Comparison to Structure of Anhydrous NiCl2
3. Experimental Section
3.1. Sample Sources
3.2. Crystallography
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kauffman, G.B. (Ed.) Coordination Chemistry: A Century of Progress; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Ibrahim, M.A.; Boeré, R.T. The copper sulfate hydration cycle. Crystal structures of CuSO4 (Chalcocyanite), CuSO4∙H2O (Poitevinite), CuSO4∙3H2O (Bonattite) and CuSO4∙5H2O (Chalcanthite) at low temperature using non-spherical atomic scattering factors. New J. Chem. 2022, 46, 5479–5488. [Google Scholar] [CrossRef]
- Scacchi, A. Dell’eriocalco e del melanotallo, nuove specie di minerali. Rend. Dell’accademia Delle Sci. Fis. E Mat. Sez. Della Soc. R. Di Napoli 1870, 9, 86–89. [Google Scholar]
- Crook, W.W., III; Jambor, J.L. Nickelbischofite, a new nickel chloride hydrate. Can. Mineral. 1979, 17, 107–109. [Google Scholar]
- N’Tsoukpoe, K.E.; Schmidt, T.; Rammelberg, H.U.; Watts, B.A.; Ruck, W.K.L. A systematic multi-step screening of numerous salt hydrates for low temperature thermochemical energy storage. Appl. Energy 2014, 124, 1–16. [Google Scholar] [CrossRef]
- Glasser, L. Thermodynamics of Inorganic Hydration and of Humidity Control, with an Extensive Database of Salt Hydrate Pairs. J. Chem. Eng. Data 2014, 59, 526–530. [Google Scholar] [CrossRef]
- Donkers, P.A.J.; Sögütoglu, L.C.; Huinink, H.P.; Fischer, H.R.; Adan, O.C.G. A review of salt hydrates for seasonal heat storage in domestic applications. Appl. Energy 2017, 199, 45–68. [Google Scholar] [CrossRef]
- Kiyabu, S.; Girard, P.; Siegel, D.J. Discovery of Salt Hydrates for Thermal Energy Storage. J. Am. Chem. Soc. 2022, 144, 21617–21627. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 4th ed.; Pearson: Harlow, UK, 2012; pp. 761–766. [Google Scholar]
- Medeiros, F.E.O.; Araujo, B.S.; Ayala, A.P. Raman spectroscopy investigation of the thermal stability of the multiferroic CuCl2 and its hydrated form. Vib. Spectr. 2018, 99, 1–6. [Google Scholar] [CrossRef]
- DeFotis, G.C.; Hampton, A.S.; Van Dongen, M.J.; Komatsu, C.H.; Benday, N.S.; Davis, C.M.; Hays, K.; Wagner, M.J. Magnetism of CuCl2·2D2O and CuCl2·2H2O, and of CuBr2·6H2O. J. Magn. Magn. Mater. 2017, 434, 23–29. [Google Scholar] [CrossRef]
- Antonijevic, S.; Persson, E. Study of water dynamics and distances in paramagnetic solids by variable-temperature two-dimensional 2H NMR sectroscopy. J. Chem. Phys. 2007, 126, 014504. [Google Scholar] [CrossRef]
- Frost, R.L.; Williams, P.A.; Kloprogge, J.T.; Martens, W. Raman spectroscopy of the copper chloride minerals nantokite, eriochalcite and claringbullite—Implications for copper corrosion. Neues Jahrb. Mineral. Monats. 2003, 2003, 433–445. [Google Scholar] [CrossRef]
- Kleinberg, R. Crystal Structure of NiCl2·6H2O at Room Temperature and 4.2°K by Neutron Diffraction. J. Chem. Phys. 1969, 50, 4690–4696. [Google Scholar] [CrossRef]
- Kleinberg, R. Magnetic Structure of NiCl2·6H2O. J. Appl. Phys. 1967, 38, 1453–1454. [Google Scholar] [CrossRef]
- Spence, R.D.; Middents, P.; ElSaffar, Z.; Kleinberg, R. A Proton Resonance Study of the Magnetic Structure of Antiferromagnetic CoCl2·6H2O, CoBr2·6H20, NiCl2·6H2O, and NiBr2·6H2O. J. Appl. Phys. 1964, 35, 854–855. [Google Scholar] [CrossRef]
- Enyashin, A.N.; Ivanovskii, A.L. Magnetic properties of NiCl2 nanostructures. Comput. Mater. Sci. 2010, 49, 782–786. [Google Scholar] [CrossRef]
- Zajnullin, O.B.; Voloshin, A.E.; Komornikov, V.A.; Manomenova, V.L.; Rudneva, E.B.; Timakov, I.S.; Kovalev, S.I. Some Properties of NiCl2·6H2O Single Crystals. Phys. Sol. State 2019, 61, 2415–2417. [Google Scholar] [CrossRef]
- Capelli, S.C.; Burgi, H.B.; Dittrich, B.; Grabowsky, S.; Jayatilaka, D. Hirshfeld atom refinement. IUCrJ 2014, 1, 361–379. [Google Scholar] [CrossRef]
- Woińska, M.; Grabowsky, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen atoms can be located accurately and precisely by x-ray crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef]
- Kleemiss, F.; Dolomanov, O.V.; Bodensteiner, M.; Peyerimhoff, N.; Midgley, L.; Bourhis, L.J.; Genoni, A.; Malaspina, L.A.; Jayatilaka, D.; Spencer, J.L.; et al. Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef]
- Bacon, G.E. Neutron Diffraction, 3rd ed.; Clarendon Press: Oxford, UK, 1975. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Crystallogr. Sect. A 2015, 71, 59–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, R.I. Recent developments in the refinement and analysis of crystal structures. Struct. Bond. 2020, 185, 43–68. [Google Scholar] [CrossRef]
- Farrugia, L.J. Accurate H-atom parameters from X-ray diffraction data. IUCrJ 2014, 1, 265–266. [Google Scholar] [CrossRef]
- Hill, N.D.D.; Lilienthal, E.; Bender, C.O.; Boeré, R.T. Accurate crystal structures of C12H9CN, C12H8(CN)2, and C16H11CN valence isomers using nonspherical atomic scattering factors. J. Org. Chem. 2022, 87, 16213–16229. [Google Scholar] [CrossRef]
- Marszaukowski, F.; Boeré, R.T.; Wohnrath, K. Frustrated and realized hydrogen bonding in 4-hydroxy-3,5-ditertbutylphenylphosphine derivatives. Cryst. Growth Des. 2022, 22, 2512–2533. [Google Scholar] [CrossRef]
- Chocolatl Torres, M.; Bernès, S.; Kuri, U.S. Refinement of K[HgI3]·H2O using non-spherical atomic form factors. Acta Crystallogr. 2021, E77, 681–685. [Google Scholar] [CrossRef]
- Chrappová, J.; Pateda, Y.R.; Rakovský, E. Synthesis and Crystal Structure Analysis of NH4[Zn(cma)(H2O)2]·H2O Using IAM and HAR Approaches. J. Chem. Crystallogr. 2022. [Google Scholar] [CrossRef]
- Harker, D. The Crystal Structure of Cupric Chloride Dihydrate CuCl2·2H2O. Z. Kristallogr. 1936, 93, 136–145. [Google Scholar] [CrossRef]
- MacGillavry, C.H.; Bijvoet, J.M. Die Kristallstruktur der Cadmium- und Quecksilber-Diamin-Dihalogenide. Z. Kristallogr. 1936, 94, 231–245. [Google Scholar] [CrossRef]
- Engberg, A. An X-ray refinement of the crystal structure of copper(II) chloride dihydrate. Acta Chem. Scand. 1970, 24, 3510–3526. [Google Scholar] [CrossRef]
- Brownstein, S.; Han, N.F.; Gabe, E.; LePage, Y. A redetermination of the crystal structure of cupric chloride dihydrate. Z. Kristallogr. 1989, 189, 13–15. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.W.; Levy, H.A. Proton Positions in CuCl2·2H2O by Neutron Diffraction. J. Chem. Phys. 1957, 26, 220. [Google Scholar] [CrossRef]
- Halcrow, M.A. Jahn-Teller distortions in transition metal compounds, and their importance in functional molecular and inorganic materials. Chem. Soc. Rev. 2013, 42, 1784–1795. [Google Scholar] [CrossRef]
- Ohtaki, H.; Yamaguchi, T.; Maeda, M. X-ray diffraction studies of the structures of hydrated divalent transition-metal ions in aqueous solution. Bull. Chem. Soc. Jpn. 1976, 49, 701–708. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Chiari, G.; Ferraris, G. The water molecule in crystalline hydrates studied by neutron diffraction. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1982, 38, 2331–2341. [Google Scholar] [CrossRef]
- Chandler, G.S.; Wajrak, M.; Khan, R.N. Neutron diffraction structures of water in crystalline hydrates of metal salts. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2015, 71, 275–284. [Google Scholar] [CrossRef]
- Burns, P.C.; Hawthorne, F.C. Tolbachite, CuCl2, the first example of Cu2+ octahedrally coordinated by Cl−. Am. Mineral 1993, 78, 187–189. [Google Scholar]
- Grazulis, S.; Chateigner, D.; Downs, R.T.; Yokochi, A.T.; Quiros, M.; Lutterotti, L.; Manakova, E.; Butkus, J.; Moeck, P.; Le Bail, A. Crystallography Open Database—An open-access collection of crystal structures. J. Appl. Crystallogr. 2009, 42, 726–729. [Google Scholar] [CrossRef]
- Ferrari, A.; Braibanti, A.; Bigliardi, G. Refinement of the crystal structure of NiCl2 and of unit-cell parameters of some anhydrous chorides of divalent metals. Acta Crystallogr. 1963, 16, 846–847. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoms 2 | Mo Kα–1a | Cu Kα–1b |
|---|---|---|
| Cu1–Cl1 | 2.2870(3) | 2.2823(4) |
| Cu1–Cl1 ii | 2.9023(3) | 2.8973(4) |
| Cu1–O1 | 1.9420(9) | 1.9414(16) |
| O1–H1 | 0.941(14) | 0.95(2) |
| Cl1–Cu1–Cl1 vi | 180.0 | 180.0 |
| Cl1 ii –Cu1–Cl1v | 180.0 | 180.0 |
| O1–Cu1–O1 iii | 180.0 | 180.0 |
| Cl1–Cu1–O1 | 90.0 | 90.0 |
| Cl1–Cu1–Cl1 v | 91.10(1) | 91.14(1) |
| Cl1 ii –Cu1–O1 | 90.0 | 90.0 |
| Cu1–O1–H1 | 123.7(10) | 126.5(16) |
| H1–O1–H1 i | 112.5(19) | 107(3) |
| D-H∙∙∙A | d(D-H)/Å | D(H-A)/Å | d(D-A)/Å | D-H-A/° | Ueq |
|---|---|---|---|---|---|
| 1 | |||||
| O1-H1∙∙∙Cl1 i a | 0.941(14) | 2.240(14) | 3.1686(7) | 169.2(14) | 0.030(4) |
| b | 0.95(2) | 2.22(2) | 3.1627(11) | 173(2) | 0.046(7) |
| c | 0.95 | 2.24 | 3.18 | 172 | — |
| 3 | |||||
| O1-H1a∙∙∙O2 ii d | 0.965(15) | 1.783(14) | 2.7401(8) | 170.5(15) | 0.030(4) |
| e | 0.94(3) | 1.81(3) | 2.7395(16) | 168(3) | 0.029(7) |
| f | 0.94(2) | 1.80(2) | 2.74(1) | 174(2) | 0.044(4) |
| g | 0.96 | 1.77 | 2.73 | 170.5 | 0.027(1) |
| O1-H1b∙∙∙Cl iii d | 0.953(14) | 2.288(15) | 3.2049(6) | 161.3(13) | 0.035(45) |
| e | 0.94(3) | 2.32(3) | 3.2013(12) | 155(2) | 0.029(7) |
| f | 0.94(2) | 2.30(2) | 3.21(1) | 164(2) | 0.043(4) |
| g | 0.97 | 2.26 | 3.19 | 161.8 | 0.029(1) |
| O2-H2a∙∙∙Cl iv d | 0.957(19) | 2.209(19) | 3.1420(10) | 164.7(18) | 0.029(5) |
| e | 1.01(5) | 2.13(5) | 3.1368(18) | 175(4) | 0.035(11) |
| f | 1.07(3) | 2.11(3) | 3.17(2) | 165(1) | 0.015(5) |
| g | 0.97(1) | 2.16(1) | 3.109(7) | 168(1) | 0.022(3) |
| O2-H2b∙∙∙O1 v d | 0.91(2) | 2.259(18) | 3.0186(10) | 140.2(6) | 0.050(7) |
| e | 0.92(4) | 2.25(3) | 3.0176(18) | 140.3(9) | 0.029(10) |
| f | 0.96 (4) | 2.27(2) | 3.06(1) | 138.3 | 0.075(1) |
| g | 0.95(1) | 2.23 | 3.007 | 138.4 | 0.025(3) |
| Atoms 2 | Mo Kα–3a | Cu Kα–3b |
|---|---|---|
| Ni–Cl | 2.3949(3) | 2.3936(5) |
| Ni–O1 | 2.0681(5) | 2.0670(11) |
| O1–H1a | 0.965(15) | 0.94(3) |
| O1–H1b | 0.953(14) | 0.94(3) |
| O2–H2a | 0.957(19) | 1.01(5) |
| O2–H2b | 0.91(2) | 0.92(4) |
| Cl–Ni–Cl i | 180.0 | 180.0 |
| O1–Ni–O1 ii | 180.0 | 180.0 |
| O1 i–Ni–O1 iii | 180.0 | 180.0 |
| Cl–Ni–O1 | 89.137(17) | 89.17(3) |
| Cl–Ni–O1 ii | 90.863(17) | 90.83(3) |
| O1–Ni–O1 i | 93.19(3) | 93.14(6) |
| O1–Ni–O1 iii | 86.81(3) | 86.86(6) |
| Ni–O1–H1a | 115.2(8) | 115.0(17)) |
| Ni–O1–H1b | 117.4(10) | 113.1(18) |
| H1a–O1–H1b | 106.9(13) | 114(2) |
| H2a–O2–H2b | 108(2) | 115(4) |
| Parameter | 1a | 1b | 3a | 3b |
|---|---|---|---|---|
| Empirical formula | Cl2CuH4O2 | Cl2CuH4O2 | Cl2H12NiO6 | Cl2H12NiO6 |
| Formula weight | 170.481 | 170.481 | 237.690 | 237.690 |
| Temperature/K | 100.01(10) | 100.01(10) | 99.98(10) | 99.99(10) |
| Crystal system | orthorhombic | orthorhombic | monoclinic | monoclinic |
| Space group | Pmna | Pmna | I2/m | I2/m |
| a/Å | 8.0553(3) | 8.0405(4) | 6.5628(4) | 6.5579(2) |
| b/Å | 3.7295(2) | 3.7238(2) | 7.0330(4) | 7.0244(3) |
| c/Å | 7.3674(3) | 7.3585(4) | 8.7326(6) | 8.7291(3) |
| β/° | 90 | 90 | 96.723(6) | 96.702(4) |
| Volume/Å3 | 221.333(17) | 220.32(2) | 400.29(4) | 399.36(3) |
| Z, Z’ | 2, 0.25 | 2, 0.25 | 2, 0.25 | 2, 0.25 |
| ρcalc/g/cm3 | 2.558 | 2.570 | 1.972 | 1.977 |
| μ/mm−1 | 5.967 | 16.849 | 3.062 | 9.551 |
| F(000) | 166.0 | 166.0 | 244.0 | 244.0 |
| Crystal size/mm3 | 0.21 × 0.04 × 0.02 | 0.16 × 0.07 × 0.06 | 0.18 × 0.1 × 0.03 | 0.23 × 0.09 × 0.04 |
| Radiation | Mo Kα (λ = 0.71073) | Cu Kα (λ = 1.54184) | Mo Kα (λ = 0.71073) | Cu Kα (λ = 1.54184) |
| 2Θ range data collect/° | 7.5 to 68.94 | 16.32 to 149.94 | 7.36 to 71.44 | 16.04 to 160.34 |
| Index ranges | −13 ≤ h ≤ 13 −6 ≤ k ≤ 6 −12 ≤ l ≤ 12 | −9 ≤ h ≤ 9 −4 ≤ k ≤ 4 −8 ≤ l ≤ 9 | −10 ≤ h ≤ 9 −11 ≤ k ≤ 11 −14 ≤ l ≤ 13 | −8 ≤ h ≤ 6 −8 ≤ k ≤ 8 −10 ≤ l ≤ 11 |
| Reflections collected | 20107 | 1200 | 4133 | 2052 |
| Independent reflections | 499 | 238 | 890 | 469 |
| Rint, Rsigma | 0.0598, 0.0170 | 0.0186, 0.0108 | 0.0318, 0.0233 | 0.0364, 0.0227 |
| Data/restraints/param | 499/6/25 | 238/0/26 | 890/0/56 | 469/24/56 |
| Goodness-of-fit on F2 | 1.016 | 1.053 | 0.996 | 1.044 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0120, wR2 = 0.0257 | R1 = 0.0133, wR2 = 0.0329 | R1 = 0.0175, wR2 = 0.0396 | R1 = 0.0276, wR2 = 0.0823 |
| Final R indexes [all data] | R1 = 0.0134, wR2 = 0.0264 | R1 = 0.0133, wR2 = 0.0329 | R1 = 0.0186, wR2 = 0.0400 | R1 = 0.0295, wR2 = 0.0843 |
| Largest diff. peak/hole/e Å−3 | 0.56/−0.42 | 0.26/−0.21 | 0.40/−0.50 | 0.46/−0.50 |
| Accession codes (CCDC) | 2238551 | 2233706 | 2238552 | 2238553 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boeré, R.T. Crystal Structures of CuCl2·2H2O (Eriochalcite) and NiCl2∙6H2O (Nickelbischofite) at Low Temperature: Full Refinement of Hydrogen Atoms Using Non-Spherical Atomic Scattering Factors. Crystals 2023, 13, 293. https://doi.org/10.3390/cryst13020293
Boeré RT. Crystal Structures of CuCl2·2H2O (Eriochalcite) and NiCl2∙6H2O (Nickelbischofite) at Low Temperature: Full Refinement of Hydrogen Atoms Using Non-Spherical Atomic Scattering Factors. Crystals. 2023; 13(2):293. https://doi.org/10.3390/cryst13020293
Chicago/Turabian StyleBoeré, René T. 2023. "Crystal Structures of CuCl2·2H2O (Eriochalcite) and NiCl2∙6H2O (Nickelbischofite) at Low Temperature: Full Refinement of Hydrogen Atoms Using Non-Spherical Atomic Scattering Factors" Crystals 13, no. 2: 293. https://doi.org/10.3390/cryst13020293
APA StyleBoeré, R. T. (2023). Crystal Structures of CuCl2·2H2O (Eriochalcite) and NiCl2∙6H2O (Nickelbischofite) at Low Temperature: Full Refinement of Hydrogen Atoms Using Non-Spherical Atomic Scattering Factors. Crystals, 13(2), 293. https://doi.org/10.3390/cryst13020293