
Carbonate Minerals’ Precipitation in the Presence of Background Electrolytes: Sr, Cs, and Li with Different Transporting Anions


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Crystallization Experiments
2.2. Chemistry of the Solutions and pH Measurement
2.3. Characterization of the Mineral Precipitates
3. Results
3.1. Evolution of Solution Chemistry
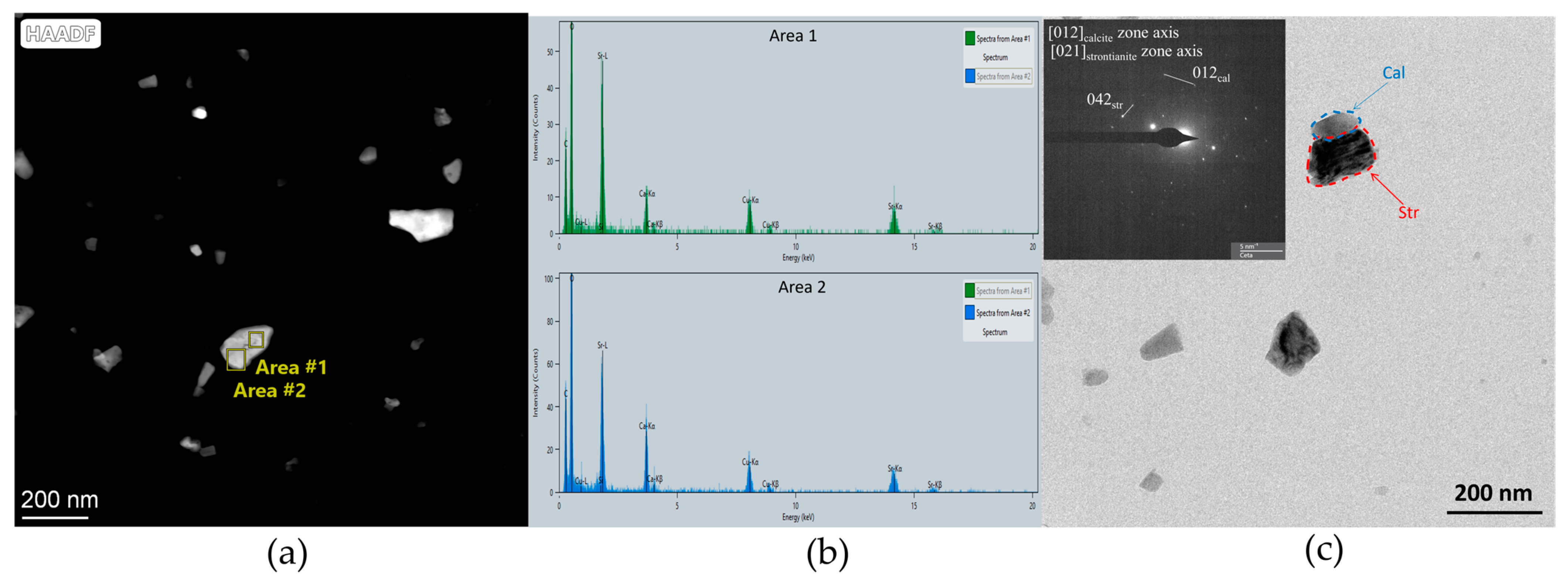
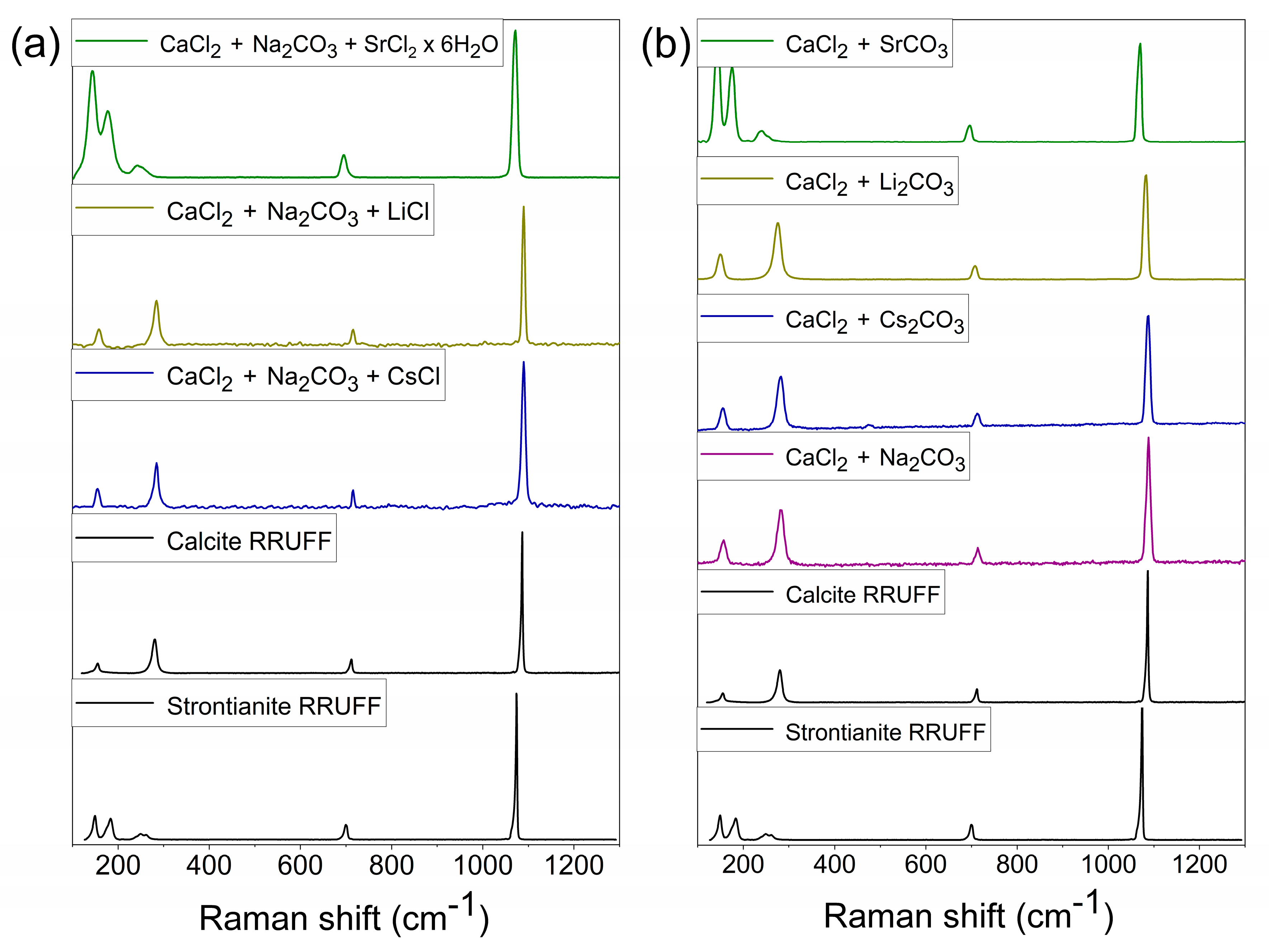
3.2. Mineral and Crystalline Characterization of the Precipitates
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Radha, A.V.; Navrotsky, A. Thermodynamics of Carbonates. Rev. Mineral. Geochem. 2013, 77, 73–121. [Google Scholar] [CrossRef]
- Falini, G.; Albeck, S.; Weiner, S.; Addadi, L. Control of Aragonite or Calcite Polymorphism by Mollusk Shell Macromolecules. Science 1996, 271, 67–69. [Google Scholar] [CrossRef]
- Christy, A.G. A Review of the Structures of Vaterite: The Impossible, the Possible, and the Likely. Cryst. Growth Des. 2017, 17, 3567–3578. [Google Scholar] [CrossRef]
- Soldati, A.L.; Jacob, D.E.; Wehrmeister, U.; Hofmeister, W. Structural characterization and chemical composition of aragonite and vaterite in freshwater cultured pearls. Mineral. Mag. 2008, 72, 579–592. [Google Scholar] [CrossRef]
- Kabalah-Amitai, L.; Mayzel, B.; Kauffmann, Y.; Fitch, A.N.; Bloch, L.; Gilbert, P.U.P.A.; Pokroy, B. Vaterite Crystals Contain Two Interspersed Crystal Structures. Science 2013, 340, 454–457. [Google Scholar] [CrossRef] [PubMed]
- McConnell, J.D.C. Vaterite from Ballycraigy, Larne, Northern Ireland. Mineral. Mag. J. Mineral. Soc. 1960, 32, 535–544. [Google Scholar] [CrossRef]
- Grasby, S. Naturally Precipitating Vaterite (μ-CaCO3) Spheres: Unusual Carbonates Formed in an Extreme Environment. Geochim. Cosmochim. Acta 2003, 67, 1659–1666. [Google Scholar] [CrossRef]
- Falkowski, P.; Scholes, R.J.; Boyle, E.; Canadell, J.; Canfield, D.; Elser, J.; Gruber, N.; Hibbard, K.; Hogberg, P.; Linder, S.; et al. The global carbon cycle: A test of our knowledge of earth as a system. Science 2000, 290, 291–296. [Google Scholar] [CrossRef]
- Meldrum, F.C. Calcium carbonate in biomineralisation and biomimetic chemistry. Int. Mater. Rev. 2003, 48, 187–224. [Google Scholar] [CrossRef]
- Cartwright, J.H.E.; Checa, A.; Gale, J.; Gebauer, D.; Sainz-Díaz, C.I. Calcium Carbonate Polyamorphism and Its Role in Biomineralization: How Many Amorphous Calcium Carbonates Are There? Angew. Chem. Int. Ed. 2012, 51, 11960–11970. [Google Scholar] [CrossRef]
- Lee, K.-M.; Park, P.-J. Estimation of the environmental credit for the recycling of granulated blast furnace slag based on LCA. Resour. Conserv. Recycl. 2005, 44, 139–151. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, C.; Elert, K.; Ševčík, R. Amorphous and crystalline calcium carbonate phases during carbonation of nanolimes: Implications in heritage conservation. Crystengcomm 2016, 18, 6594–6607. [Google Scholar] [CrossRef]
- Butler, M.F.; Glaser, N.; Weaver, A.C.; Kirkland, M.; Heppenstall-Butler, M. Calcium Carbonate Crystallization in the Presence of Biopolymers. Cryst. Growth Des. 2006, 6, 781–794. [Google Scholar] [CrossRef]
- Kirboga, S.; Öner, M. Investigation of calcium carbonate precipitation in the presence of carboxymethyl inulin. Crystengcomm 2013, 15, 3678–3686. [Google Scholar] [CrossRef]
- Buhmann, D.; Dreybrodt, W. Calcite dissolution kinetics in the system H2O CO2 CaCO3 with participation of foreign ions. Chem. Geol. 1987, 64, 89–102. [Google Scholar] [CrossRef]
- Burgos-Cara, A.; Putnis, C.V.; Rodriguez-Navarro, C.; Ruiz-Agudo, E. Hydration Effects on the Stability of Calcium Carbonate Pre-Nucleation Species. Minerals 2017, 7, 126. [Google Scholar] [CrossRef]
- Kowacz, M.; Putnis, A. The effect of specific background electrolytes on water structure and solute hydration: Consequences for crystal dissolution and growth. Geochim. Cosmochim. Acta 2008, 72, 4476–4487. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Urosevic, M.; Putnis, C.V.; Rodríguez-Navarro, C.; Cardell, C.; Putnis, A. Ion-specific effects on the kinetics of mineral dissolution. Chem. Geol. 2011, 281, 364–371. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Kowacz, M.; Putnis, C.; Putnis, A. The role of background electrolytes on the kinetics and mechanism of calcite dissolution. Geochim. Cosmochim. Acta 2010, 74, 1256–1267. [Google Scholar] [CrossRef]
- Collins, K. Charge density-dependent strength of hydration and biological structure. Biophys. J. 1997, 72, 65–76. [Google Scholar] [CrossRef]
- De Yoreo, J.J. Principles of Crystal Nucleation and Growth. Rev. Mineral. Geochem. 2003, 54, 57–93. [Google Scholar] [CrossRef]
- Fernandez-Gonzalez, A.; Lozano-Letellier, A.; Fernandez, B. The Influence of Aqueous Se(IV) on the Stability of Different CaCO3 Polymorphs Precipitated under Ambient Conditions. Minerals 2021, 11, 1238. [Google Scholar] [CrossRef]
- Paquette, J.; Reeder, R.J. Relationship between surface structure, growth mechanism, and trace element incorporation in calcite. Geochim. Cosmochim. Acta 1995, 59, 735–749. [Google Scholar] [CrossRef]
- Prieto, M.; Fernández-González, A.; Putnis, A.; Fernández-Díaz, L. Nucleation, growth, and zoning phenomena in crystallizing (Ba,Sr)CO3, Ba(SO4,CrO4), (Ba,Sr)SO4, and (Cd,Ca)CO3 solid solutions from aqueous solutions. Geochim. Cosmochim. Acta 1997, 61, 3383–3397. [Google Scholar] [CrossRef]
- Ebensperger, A.; Maxwell, P.; Moscoso, C. The lithium industry: Its recent evolution and future prospects. Resour. Policy 2005, 30, 218–231. [Google Scholar] [CrossRef]
- Bolan, N.; Hoang, S.A.; Tanveer, M.; Wang, L.; Bolan, S.; Sooriyakumar, P.; Robinson, B.; Wijesekara, H.; Wijesooriya, M.; Keerthanan, S.; et al. From mine to mind and mobiles–Lithium contamination and its risk management. Environ. Pollut. 2021, 290, 118067. [Google Scholar] [CrossRef]
- Greim, P.; Solomon, A.A.; Breyer, C. Assessment of lithium criticality in the global energy transition and addressing policy gaps in transportation. Nat. Commun. 2020, 11, 4570. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Hu, H.; Zhang, A.C.; Huang, X.-L.; Ni, P. Pollucite and the cesium-dominant analogue of polylithionite as expressions of extreme cs enrichment in the yichun topaz–lepidolite granite, southern china. Can. Mineral. 2004, 42, 883–896. [Google Scholar] [CrossRef]
- Butterman, W.C.; Brooks, W.E.; Reese, R.G. Mineral Commodity Profiles: Cesium; USGS: Reston, VA, USA, 2005. [Google Scholar] [CrossRef]
- Chen, W.; Zhu, Y. Preparation of strontium carbonate from celestite. Miner. Process. Extr. Met. 2000, 109, 1–5. [Google Scholar] [CrossRef]
- Ober, J.A. Mineral Commodity Summaries 2016; USGS: Reston, VA, USA, 2016. [Google Scholar]
- Lenihan, J.M.A. Strontium Metabolism. Nature 1966, 211, 572–573. [Google Scholar] [CrossRef]
- Kaushal, S.S.; Groffman, P.M.; Likens, G.E.; Belt, K.T.; Stack, W.P.; Kelly, V.R.; Band, L.E.; Fisher, G.T. Increased salinization of fresh water in the northeastern United States. Proc. Natl. Acad. Sci. USA 2005, 102, 13517–13520. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, D.A. Water Resources Bulletin. Am. Water Resour. Assoc. 1986, 22, 57–65. [Google Scholar] [CrossRef]
- Last, G.V.; Schmick, M.T. Identification and Selection of Major Carbon Dioxide Stream Compositions GV Last MT Schmick; No. PNNL-20493; Pacific Northwest National Lab. (PNNL): Richland, WA, USA, 2011. [Google Scholar]
- Sazali, Y.; Sazali, W.; Ibrahim, J.; Dindi, M.; Graham, G.; Gödeke, S. Investigation of high temperature, high pressure, scaling and dissolution effects for Carbon Capture and Storage at a high CO2 content carbonate gas field offshore Malaysia. J. Pet. Sci. Eng. 2018, 174, 599–606. [Google Scholar] [CrossRef]
- Yoon, I.-S.; Chang, C.-H. Effect of Chloride on Electrical Resistivity in Carbonated and Non-Carbonated Concrete. Appl. Sci. 2020, 10, 6272. [Google Scholar] [CrossRef]
- Liu, W.; Cao, F.; Chen, A.; Chang, L.; Zhang, J.; Cao, C. Effect of Chloride Ion Concentration on Electrochemical Behavior and Corrosion Product of AM60 Magnesium Alloy in Aqueous Solutions. Corrosion 2012, 68, 45001. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo1, C.A.J. User’s Guide to Phreeqc (Version 2) A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculation; USGS: Reston, VA, USA, 1999. [Google Scholar]
- Wang, X.; Ye, Y.; Wu, X.; Smyth, J.R.; Yang, Y.; Zhang, Z.; Wang, Z. High-temperature Raman and FTIR study of aragonite-group carbonates. Phys. Chem. Miner. 2019, 46, 51–62. [Google Scholar] [CrossRef]
- Dufresne, W.J.; Rufledt, C.J.; Marshall, C.P. Raman spectroscopy of the eight natural carbonate minerals of calcite structure. J. Raman Spectrosc. 2018, 49, 1999–2007. [Google Scholar] [CrossRef]
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Hong, J.; Heo, S.J.; Singh, P. Water mediated growth of oriented single crystalline SrCO3 nanorod arrays on strontium compounds. Sci. Rep. 2021, 11, 3368. [Google Scholar] [CrossRef]
- Graf, D.L. Crystallographic Tables for the Rhombohedral Carbonates. Am. Mineral. 1961, 46, 1283–1316. [Google Scholar]
- de Villiers, D.M. Crystal Structures of Aragonite, Strontianite, and Witherite. Am. Mineral. 1971, 56, 758–767. [Google Scholar]
- Rodriguez-Navarro, C.; Jimenez-Lopez, C.; Rodriguez-Navarro, A.; González-Muñoz, M.T.; Rodriguez-Gallego, M. Bacterially mediated mineralization of vaterite. Geochim. Cosmochim. Acta 2007, 71, 1197–1213. [Google Scholar] [CrossRef]
- Alia, J.M.; Diaz De Mera, Y.; Edwards, H.G.M.; Gonzsilez Martin, P.; Andres, L.S. FT-Raman and Infrared Spectroscopic Study of Aragonite-Strontianite (Ca,Sr) Solid Solution. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1997, 53, 2347–2362. [Google Scholar] [CrossRef]
- Donnelly, F.C.; Purcell-Milton, F.; Framont, V.; Cleary, O.; Dunne, P.W.; Gun’Ko, Y.K. Synthesis of CaCO3nano- and micro-particles by dry ice carbonation. Chem. Commun. 2017, 53, 6657–6660. [Google Scholar] [CrossRef] [PubMed]
- DeCarlo, T.M. Characterizing coral skeleton mineralogy with Raman spectroscopy. Nat. Commun. 2018, 9, 5325. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Liu, L.-G. Post-aragonite phase transitions in strontianite and cerussite—A high pressure Raman spectroscopic study. J. Phys. Chem. Solids 1997, 58, 977–987. [Google Scholar] [CrossRef]
- Chang, R.; Kim, S.; Lee, S.; Choi, S.; Kim, M.; Park, Y. Calcium Carbonate Precipitation for CO2 Storage and Utilization: A Review of the Carbonate Crystallization and Polymorphism. Front. Energy Res. 2017, 5, 17. [Google Scholar] [CrossRef]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable Prenucleation Calcium Carbonate Clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef]
- Wada, N.; Yamashita, K.; Umegaki, T. Effects of divalent cations upon nucleation, growth and transformation of calcium carbonate polymorphs under conditions of double diffusion. J. Cryst. Growth 1995, 148, 297–304. [Google Scholar] [CrossRef]
- Mergelsberg, S.T.; Riechers, S.L.; Graham, T.R.; Prange, M.P.; Kerisit, S.N. Effect of Cd on the Nucleation and Transformation of Amorphous Calcium Carbonate. Cryst. Growth Des. 2021, 21, 3384–3393. [Google Scholar] [CrossRef]
- Liendo, F.; Arduino, M.; Deorsola, F.A.; Bensaid, S. Factors controlling and influencing polymorphism, morphology and size of calcium carbonate synthesized through the carbonation route: A review. Powder Technol. 2022, 398, 117050. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Fernández-González, A.; Prieto, M. The role of sulfate groups in controlling CaCO3 polymorphism. Geochim. Cosmochim. Acta 2010, 74, 6064–6076. [Google Scholar] [CrossRef]
- Mlinarić, N.M.; Kontrec, J.; Džakula, B.N.; Falini, G.; Kralj, D. Role of Hydrodynamics, Li+ Addition and Transformation Kinetics on the Formation of Plate-Like {001} Calcite Crystals. Crystals 2021, 11, 250. [Google Scholar] [CrossRef]
- Pastero, L.; Costa, E.; Bruno, M.; Rubbo, M.; Sgualdino, G.; Aquilano, D. Morphology of Calcite (CaCO3) Crystals Growing from Aqueous Solutions in the Presence of Li+ Ions. Surface Behavior of the {0001} Form. Cryst. Growth Des. 2004, 4, 485–490. [Google Scholar] [CrossRef]
- Aquilano, D.; Bruno, M.; Pastero, L. Impurity Effects on Habit Change and Polymorphic Transitions in the System: Aragonite–Calcite–Vaterite. Cryst. Growth Des. 2020, 20, 2497–2507. [Google Scholar] [CrossRef]
- Matijaković, N.; Magnabosco, G.; Scarpino, F.; Fermani, S.; Falini, G.; Kralj, D. Synthesis and Adsorbing Properties of Tabular {001} Calcite Crystals. Crystals 2019, 9, 16. [Google Scholar] [CrossRef]
- Füger, A.; Konrad, F.; Leis, A.; Dietzel, M.; Mavromatis, V. Effect of growth rate and pH on lithium incorporation in calcite. Geochim. Cosmochim. Acta 2019, 248, 14–24. [Google Scholar] [CrossRef]
- Zhang, D.-K.; Zhou, X.-Q.; Liu, C.-L.; Lu, G.-M. Crystallization of calcium carbonate from lithium-containing brines. J. Cryst. Growth 2020, 556, 125989. [Google Scholar] [CrossRef]
- Devriendt, L.; Mezger, E.; Olsen, E.; Watkins, J.; Kaczmarek, K.; Nehrke, G.; de Nooijer, L.; Reichart, G.-J. Sodium incorporation into inorganic CaCO3 and implications for biogenic carbonates. Geochim. Cosmochim. Acta 2021, 314, 294–312. [Google Scholar] [CrossRef]
- Wehrmeister, U.; Soldati, A.L.; Jacob, D.E.; Häger, T.; Hofmeister, W. Raman spectroscopy of synthetic, geological and biological vaterite: A Raman spectroscopic study. J. Raman Spectrosc. 2009, 41, 193–201. [Google Scholar] [CrossRef]
- Yin, X.; Wang, X.; Wu, H.; Ohnuki, T.; Takeshita, K. Enhanced desorption of cesium from collapsed interlayer regions in vermiculite by hydrothermal treatment with divalent cations. J. Hazard. Mater. 2017, 326, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.K.; Kuwata, M.; Ito, A.; Umita, T. Effect of pH and Chemical Composition of Solution on Sorption and Retention of Cesium by Feldspar, Illite, and Zeolite as Cesium Sorbent from Landfill Leachate. Soil Sci. 2017, 182, 1. [Google Scholar] [CrossRef]
- Cornell, R.M. Adsorption of cesium on minerals: A review. J. Radioanal. Nucl. Chem. 1993, 171, 483–500. [Google Scholar] [CrossRef]
- Curti, E. Coprecipitation of radionuclides with calcite: Estimation of partition coefficients based on a review of laboratory investigations and geochemical data. Appl. Geochem. 1999, 14, 433–445. [Google Scholar] [CrossRef]
- Fujikawa, Y.; Fukui, M. Radionuclide Sorption to Rocks and Minerals: Effects of pH and Inorganic Anions. Part 1. Sorption of Cesium, Cobalt, Strontium and Manganese. Radiochim. Acta 1997, 76, 153–162. [Google Scholar] [CrossRef]
- Kim, Y.; Kwon, S.; Roh, Y. Effect of Divalent Cations (Cu, Zn, Pb, Cd, and Sr) on Microbially Induced Calcium Carbonate Precipitation and Mineralogical Properties. Front. Microbiol. 2021, 12, 646748. [Google Scholar] [CrossRef]
- Sunagawa, I.; Takahashi, Y.; Imai, H. Strontium and aragonite-calcite precipitation. J. Mineral. Pet. Sci. 2007, 102, 174–181. [Google Scholar] [CrossRef]
- Wasylenki, L.E.; Dove, P.M.; Wilson, D.S.; De Yoreo, J.J. Nanoscale effects of strontium on calcite growth: An in situ AFM study in the absence of vital effects. Geochim. Cosmochim. Acta 2005, 69, 3017–3027. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | Sample | Calcite | Vaterite | Aragonite | Strontianite |
|---|---|---|---|---|---|
| 24 h | CaCl2 + Na2CO3 | 79.3 | 20.7 | - | - |
| CaCl2 + Na2CO3 + CsCl | 100 | - | - | - | |
| CaCl2 + Cs2CO3 | 100 | - | - | - | |
| CaCl2 + Na2CO3 + LiCl | 8.1 | 91.9 | - | - | |
| CaCl2 + Li2CO3 | 3.8 | 96.2 | - | - | |
| CaCl2 + Na2CO3 + SrCl2 | - | - | - | 100 | |
| CaCl2 + SrCO3 | - | - | 4.8 | 95.2 | |
| 48 h | CaCl2 + Na2CO3 | 100 | - | - | - |
| CaCl2 + Na2CO3 + CsCl | 100 | - | - | - | |
| CaCl2 + Cs2CO3 | 100 | - | - | - | |
| CaCl2 + Na2CO3 + LiCl | 16.3 | 83.7 | - | - | |
| CaCl2 + Li2CO3 | 24.6 | 75.4 | - | - | |
| CaCl2 + Na2CO3 + SrCl2 | 3.9 | - | - | 96.1 | |
| CaCl2 + SrCO3 | - | - | - | 100 | |
| 1 week | CaCl2 + Na2CO3 | 100 | - | - | - |
| CaCl2 + Na2CO3 + CsCl | 100 | - | - | - | |
| CaCl2 + Cs2CO3 | 100 | - | - | - | |
| CaCl2 + Na2CO3 + LiCl | 40.9 | 59.1 | - | - | |
| CaCl2 + Li2CO3 | 63.1 | 36.9 | - | - | |
| CaCl2 + Na2CO3 + SrCl2 | 5.6 | - | - | 94.4 | |
| CaCl2 + SrCO3 | - | - | - | 100 | |
| 1 month | CaCl2 + Na2CO3 | 100 | - | - | - |
| CaCl2 + Na2CO3 + CsCl | 100 | - | - | - | |
| CaCl2 + Cs2CO3 | 100 | - | - | - | |
| CaCl2 + Na2CO3 + LiCl | 100 | - | - | - | |
| CaCl2 + Li2CO3 | 100 | - | - | - | |
| CaCl2 + Na2CO3 + SrCl2 | 7.5 | - | - | 92.5 | |
| CaCl2 + SrCO3 | - | - | - | 100 |
| 24 h | 48 h | 168 h | 720 h | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mineral | Experiment | a | c | a | c | a | c | a | c | ||||
| Calcite rhombohedral | CaCl2 + Na2CO3 | 4.9893 (3) | 17.0545 (1) | 4.9909 (9) | 17.0634 (5) | 4.9913 (6) | 17.0652 (1) | 4.9910 (6) | 17.0589 (1) | ||||
| CaCl2 + Na2CO3 + CsCl | 4.9903 (4) | 17.0625 (1) | 4.9912 (2) | 17.0663 (1) | 4.9910 (6) | 17.0647 (8) | 4.9902 (7) | 17.0651 (6) | |||||
| CaCl2 + Cs2CO3 | 4.9902 (1) | 17.0601 (7) | 4.9907 (2) | 17.0635 (9) | 4.9911 (8) | 17.0633 (3) | 4.9924 (9) | 17.0591 (7) | |||||
| CaCl2 + Na2CO3 + LiCl | 4.9902 (1) | 17.0648 (7) | 4.9910 (5) | 17.0670 (9) | 4.9915 (1) | 17.0702 (1) | 4.9894 (5) | 17.0649 (9) | |||||
| CaCl2 + Li2CO3 | 4.9928 (2) | 17.0729 (2) | 4.9901 (5) | 17.0627 (4) | 4.9909 (4) | 17.0658 (5) | 4.9913 (2) | 17.0646 (3) | |||||
| CaCl2 + Na2CO3 + SrCl2 | 4.9798 (4) | 17.0838 (3) | 4.9878 (3) | 17.0755 (5) | 4.9880 (6) | 17.0679 (2) | |||||||
| CaCl2 + SrCO3 | |||||||||||||
| Vaterite rhombohedral | CaCl2 + Na2CO3 | 4.1284 (9) | 8.4665 (4) | ||||||||||
| CaCl2 + Na2CO3 + LiCl | 4.1307 (3) | 8.4771 (9) | 4.1278 (3) | 8.4706 (1) | 4.1296 (5)1 | 8.4712 (6) | |||||||
| CaCl2 + Li2CO3 | 4.1295 (9) | 8.4751 (8) | 4.1299 (5) | 8.4745 (1) | 4.1300 (7) | 8.4730 (1) | |||||||
| a | b | c | a | b | c | a | b | c | a | b | c | ||
| Strontianite orthorhombic | CaCl2 + SrCO3 | 6.0336 (1) | 5.1075 (2) | 8.4249 (1) | 6.0322 (7) | 5.1068 (7) | 8.4217(6) | 6.0324 (5) | 5.1070 (2) | 8.4213(4) | 6.0328 (6) | 5.1074 (1) | 8.4213 (1) |
| CaCl2 + Na2CO3 + SrCl2 | 5.8708 (6) | 4.8469 (3) | 8.5898 (9) | 5.8743 (7) | 4.9647 (4) | 8.3744(2) | 5.9563 (1) | 5.0449 (4) | 8.3225(4) | 5.9478 (5) | 5.0175 (5) | 8.3908 (9) | |
| Aragonite orthorhombic | CaCl2 + Na2CO3 + SrCl2 | 6.0275 (6) | 4.7251 (7) | 7.8750 (8) | |||||||||
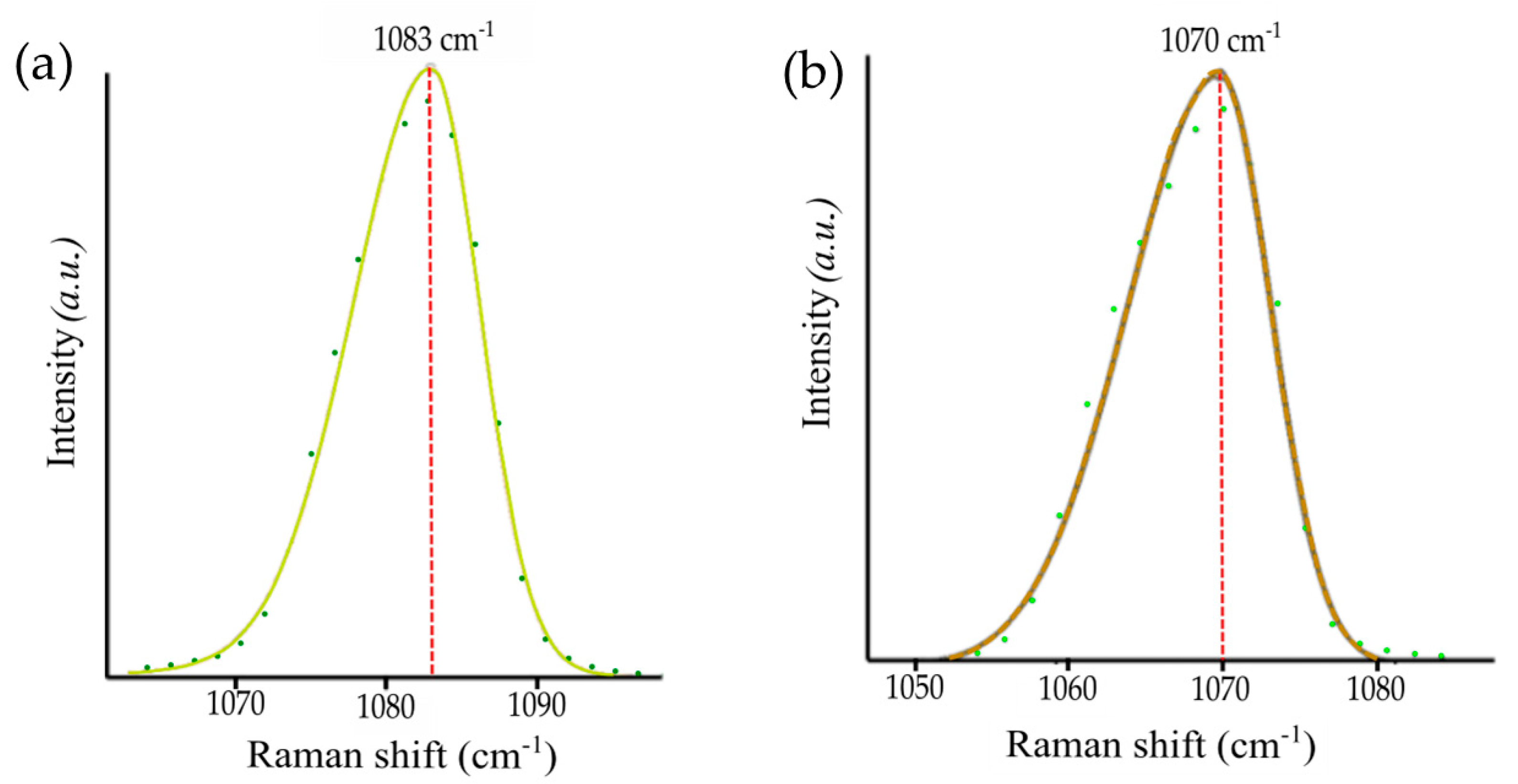
| Sample | Calcite ν1 | Strontianite ν1 | ||
|---|---|---|---|---|
| Band Position | FWHM | Band Position | FWHM | |
| CaCl2 + Na2CO3 | 1083.0 (7) | 10.3 (8) | - | - |
| CaCl2 + Na2CO3 + CsCl | 1083.4 (1) | 11.0 (4) | - | - |
| CaCl2 + Cs2CO3 | 1081.7 (3) | 10.5 (2) | - | - |
| CaCl2 + Na2CO3 + LiCl | 1086.0 (1) | 10.9 (1) | - | - |
| CaCl2 + Li2CO3 | 1084.3 (4) | 10.7 (8) | - | - |
| CaCl2 + Na2CO3 + SrCl2 | - | - | 1071.1 (6) | 12.5 (1) |
| CaCl2 + SrCO3 | - | - | 1070.0 (1) | 10.8 (3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marin-Troya, P.; Espinosa, C.; Monasterio-Guillot, L.; Alvarez-Lloret, P. Carbonate Minerals’ Precipitation in the Presence of Background Electrolytes: Sr, Cs, and Li with Different Transporting Anions. Crystals 2023, 13, 796. https://doi.org/10.3390/cryst13050796
Marin-Troya P, Espinosa C, Monasterio-Guillot L, Alvarez-Lloret P. Carbonate Minerals’ Precipitation in the Presence of Background Electrolytes: Sr, Cs, and Li with Different Transporting Anions. Crystals. 2023; 13(5):796. https://doi.org/10.3390/cryst13050796
Chicago/Turabian StyleMarin-Troya, Pedro, Carlos Espinosa, Luis Monasterio-Guillot, and Pedro Alvarez-Lloret. 2023. "Carbonate Minerals’ Precipitation in the Presence of Background Electrolytes: Sr, Cs, and Li with Different Transporting Anions" Crystals 13, no. 5: 796. https://doi.org/10.3390/cryst13050796
APA StyleMarin-Troya, P., Espinosa, C., Monasterio-Guillot, L., & Alvarez-Lloret, P. (2023). Carbonate Minerals’ Precipitation in the Presence of Background Electrolytes: Sr, Cs, and Li with Different Transporting Anions. Crystals, 13(5), 796. https://doi.org/10.3390/cryst13050796