
Electronic Properties, Linear and Nonlinear Performance of KAgCh (Ch = S, Se) Compounds: A First-Principles Study

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Computational Method
3. Results and Discussion
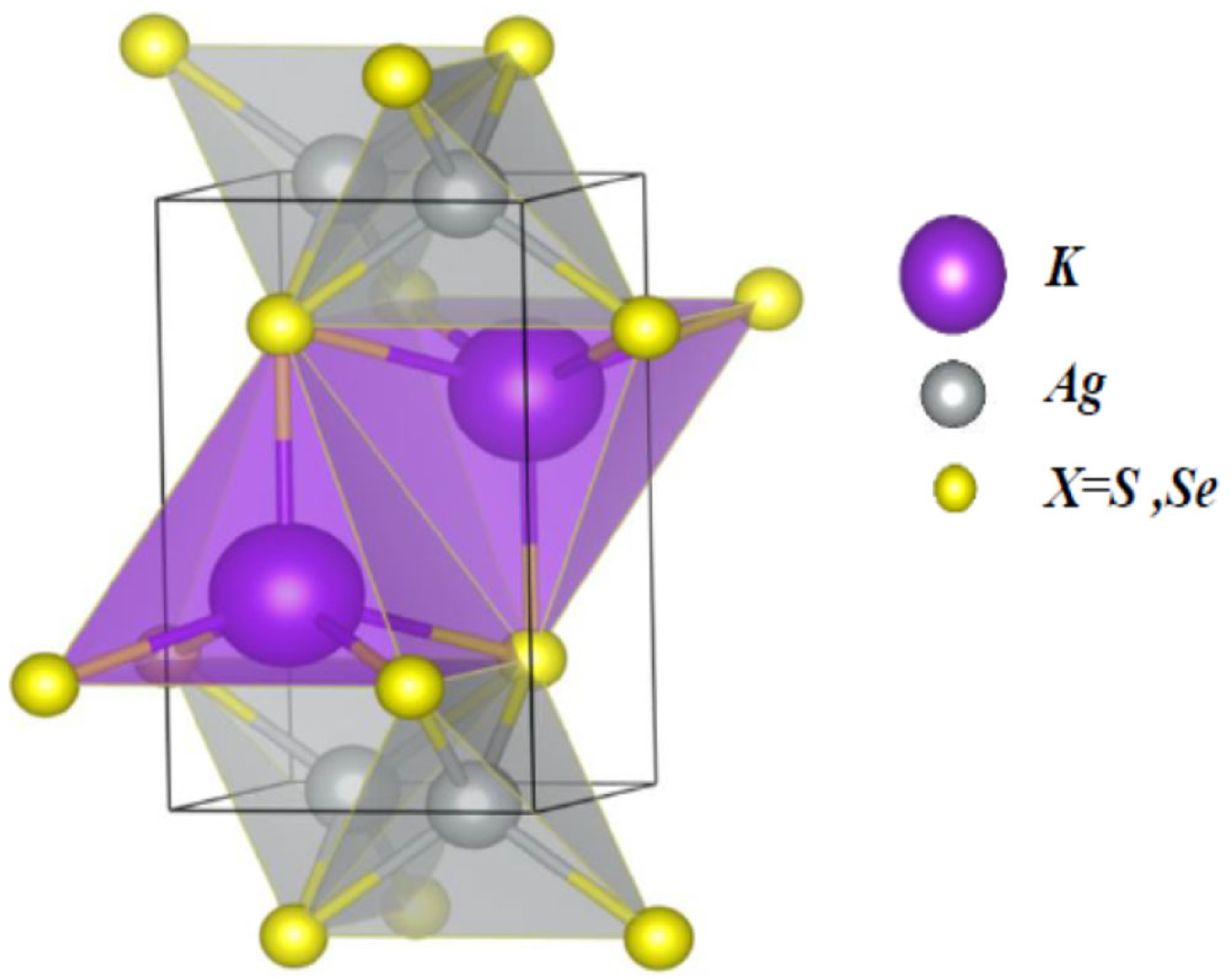
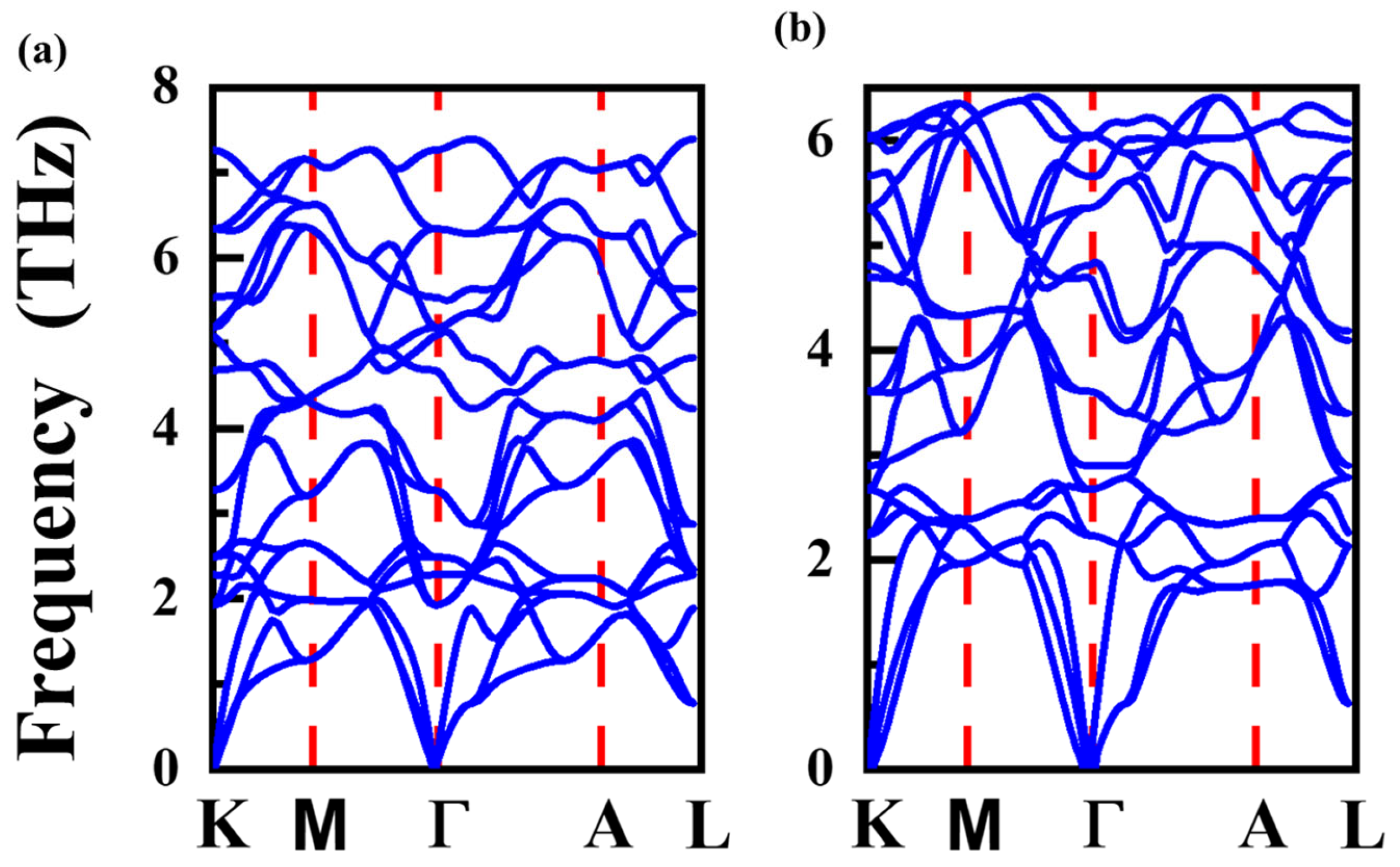
3.1. Structural Properties
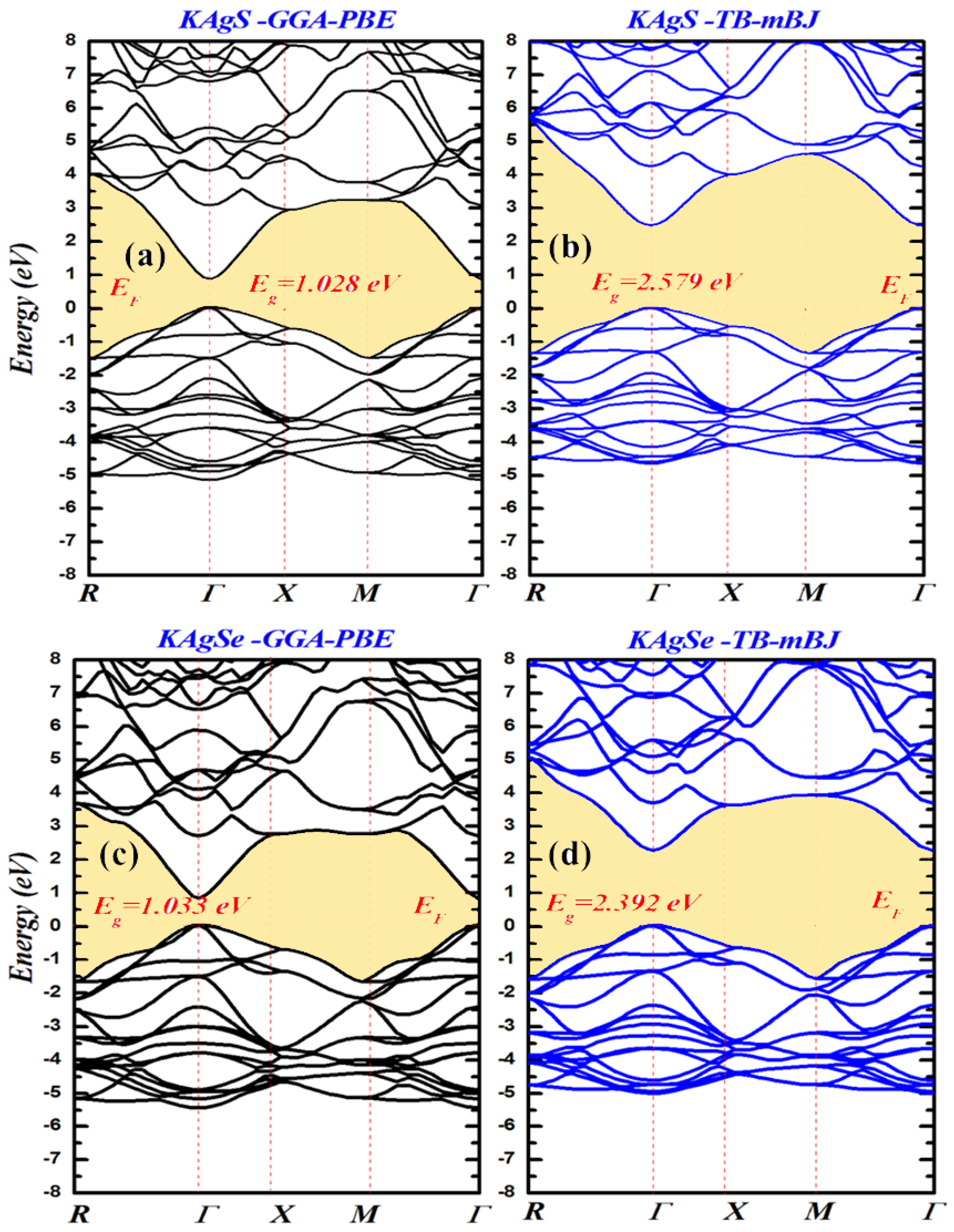
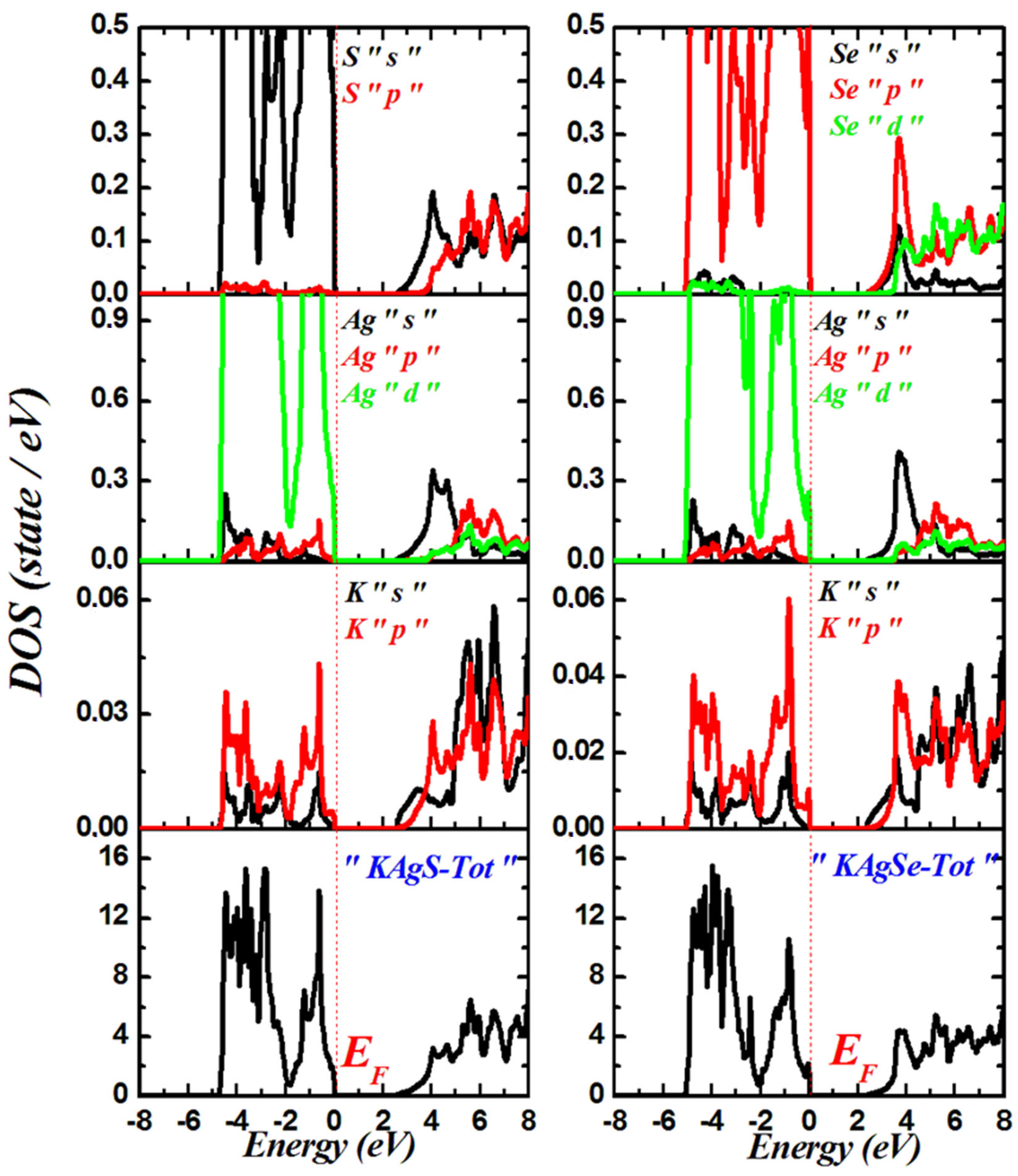
3.2. Electronic Properties
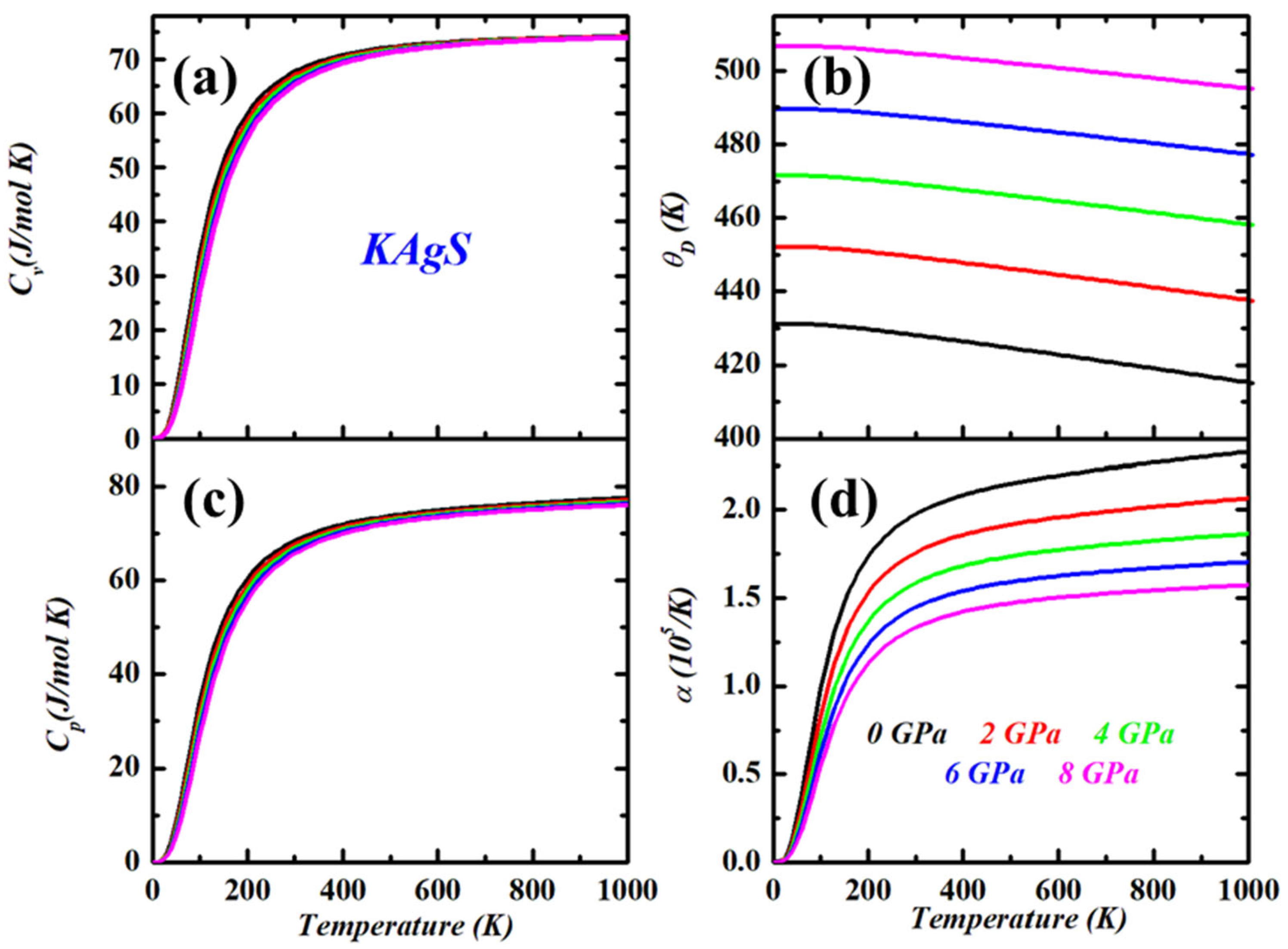
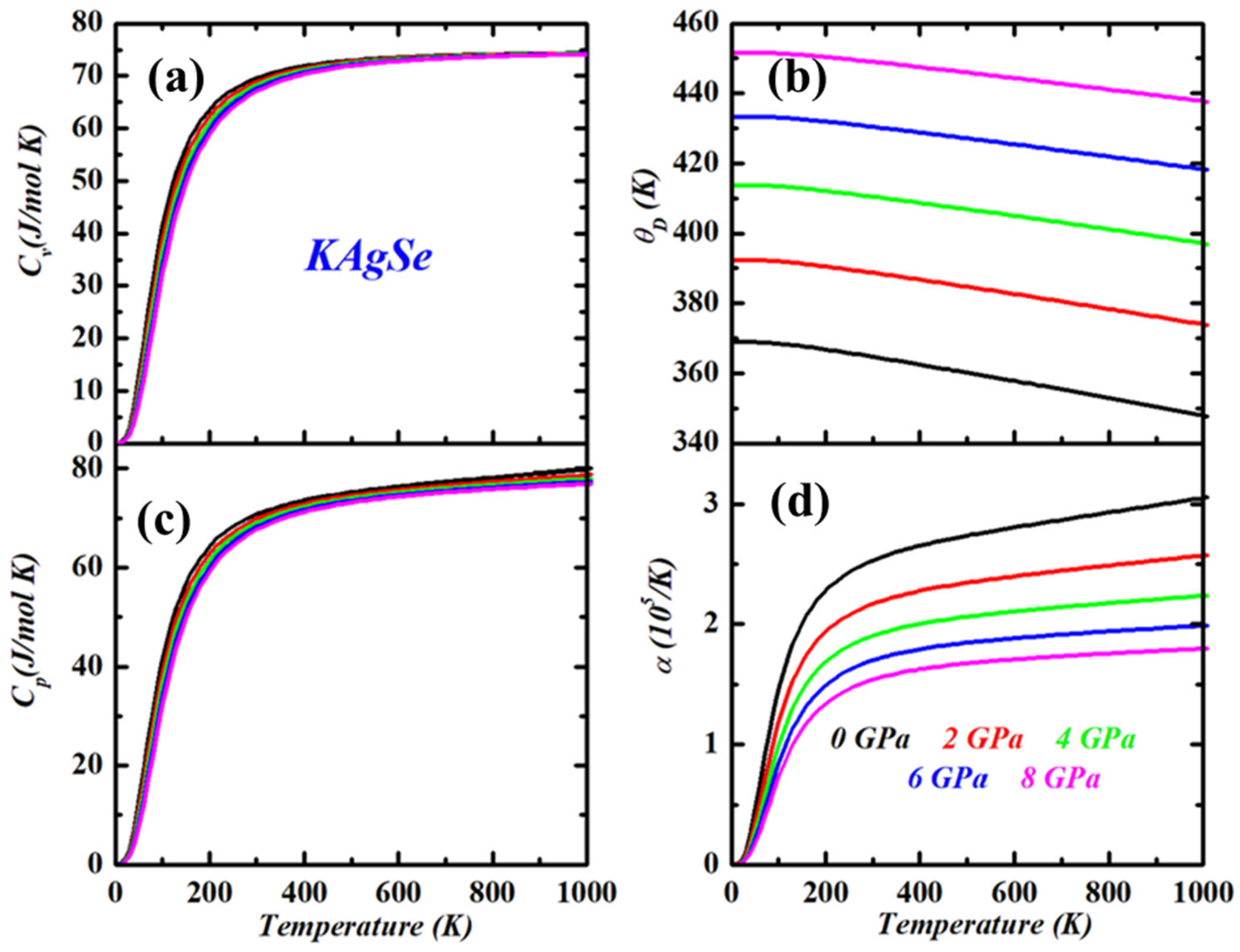
3.3. Thermodynamic Properties
3.4. Optical Properties
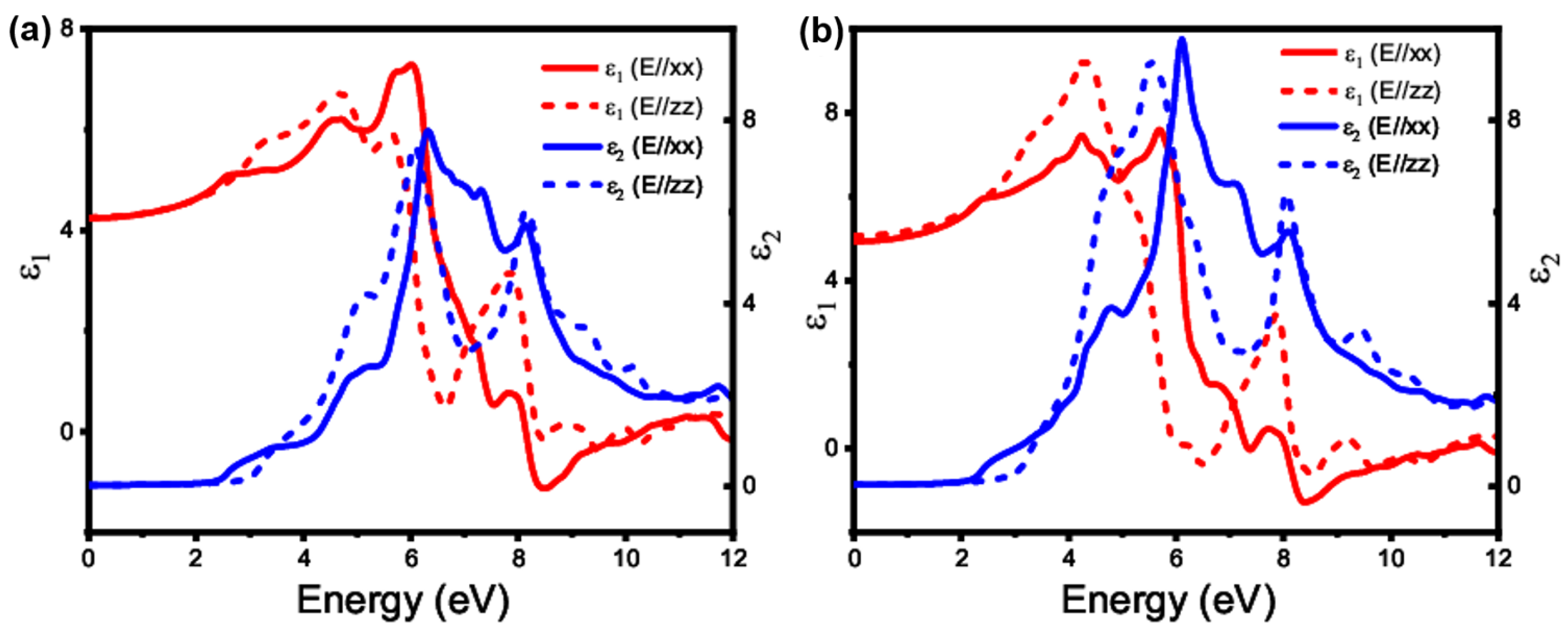
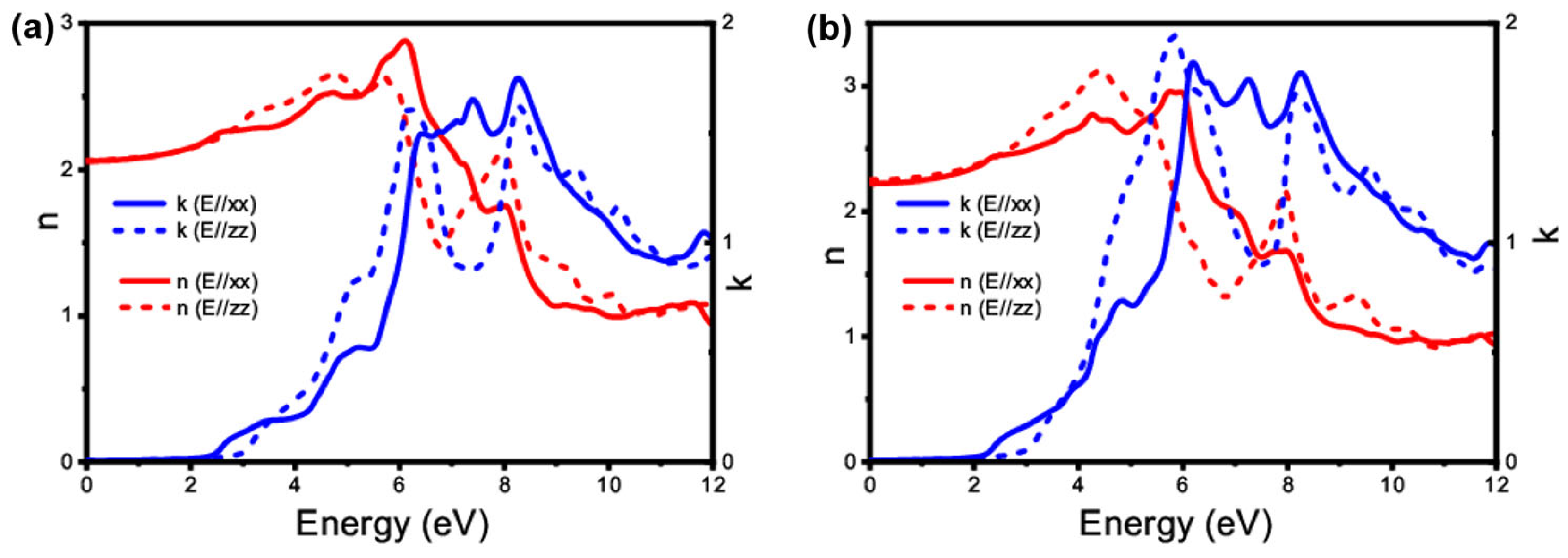
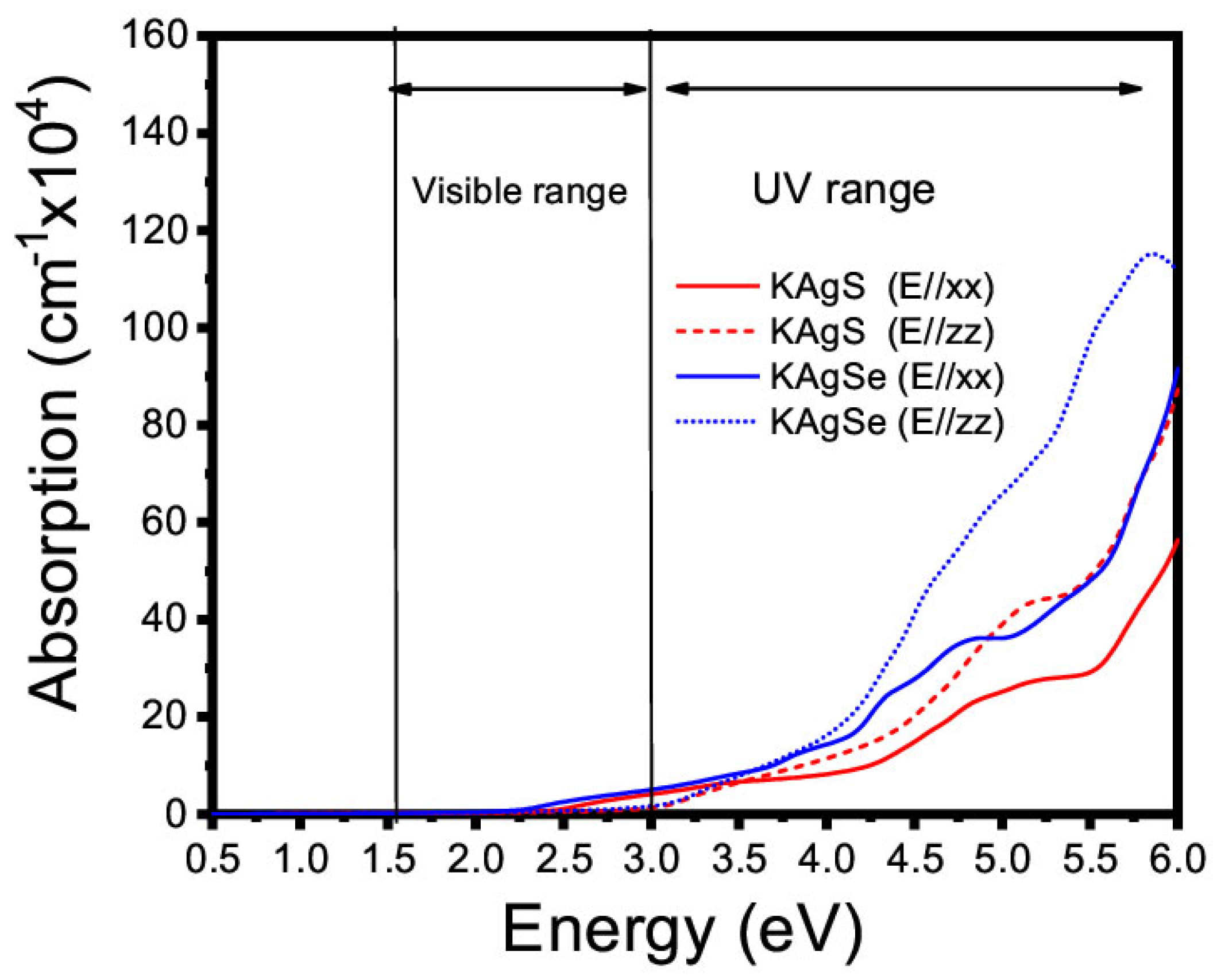
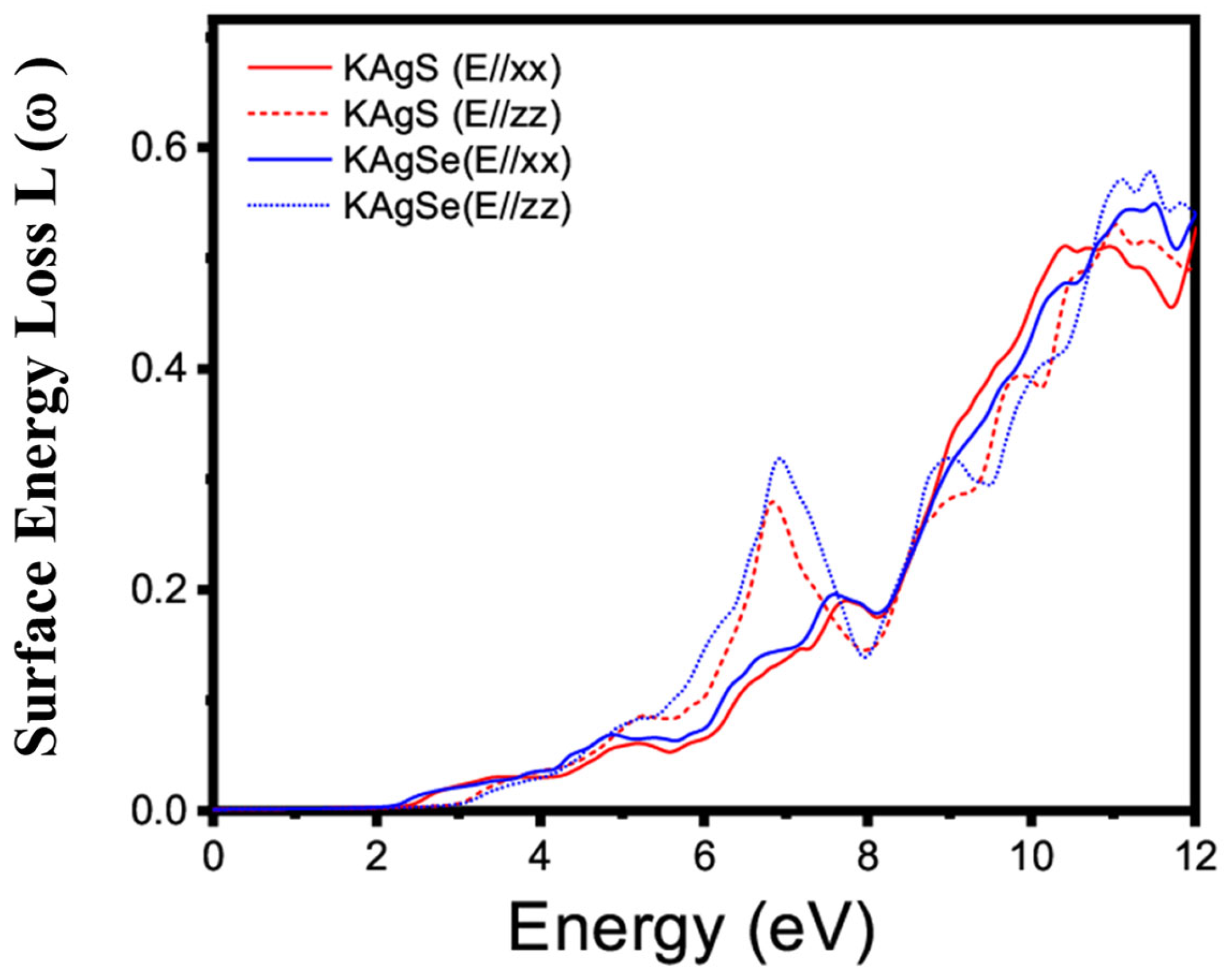
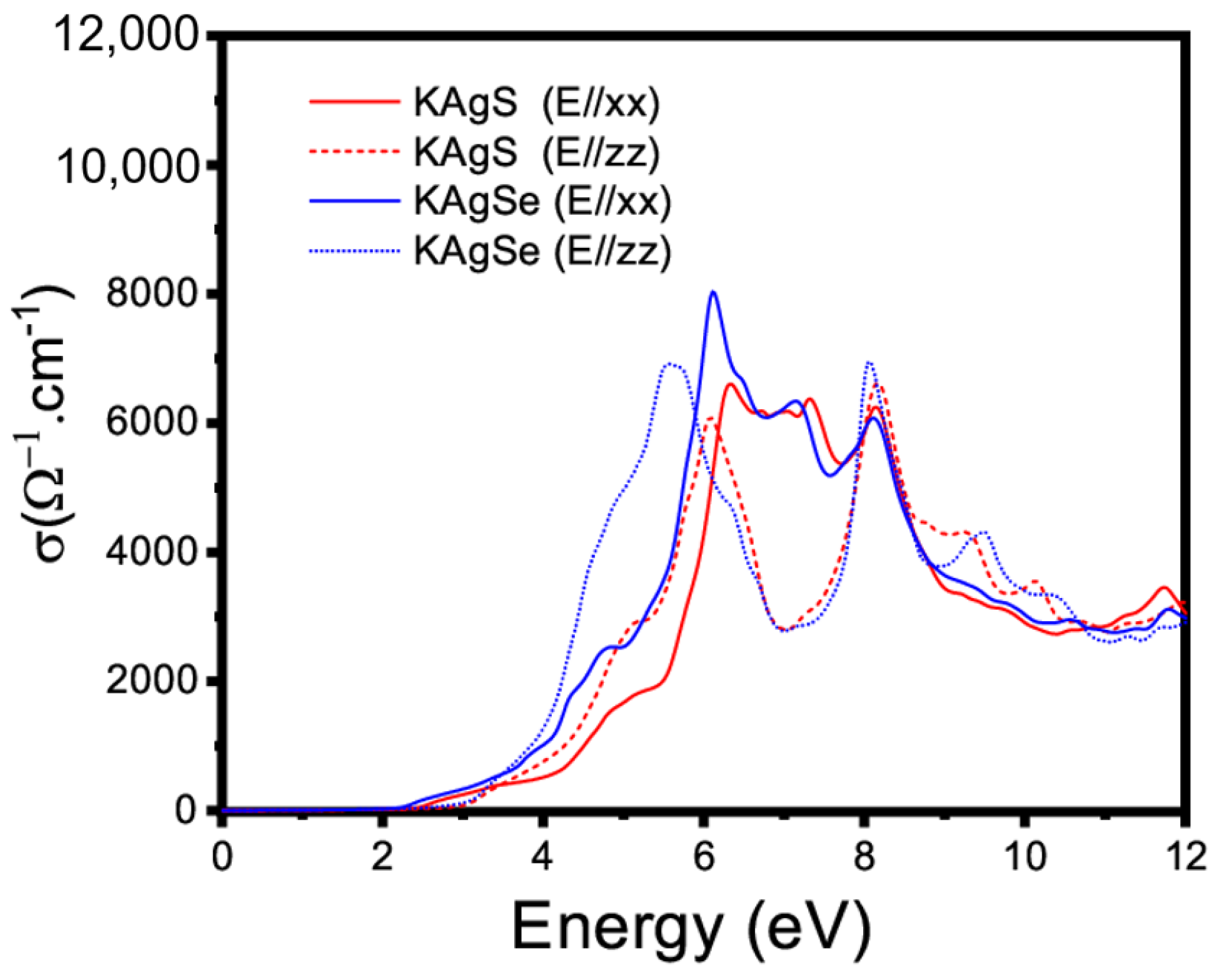
3.4.1. Linear Optical Property
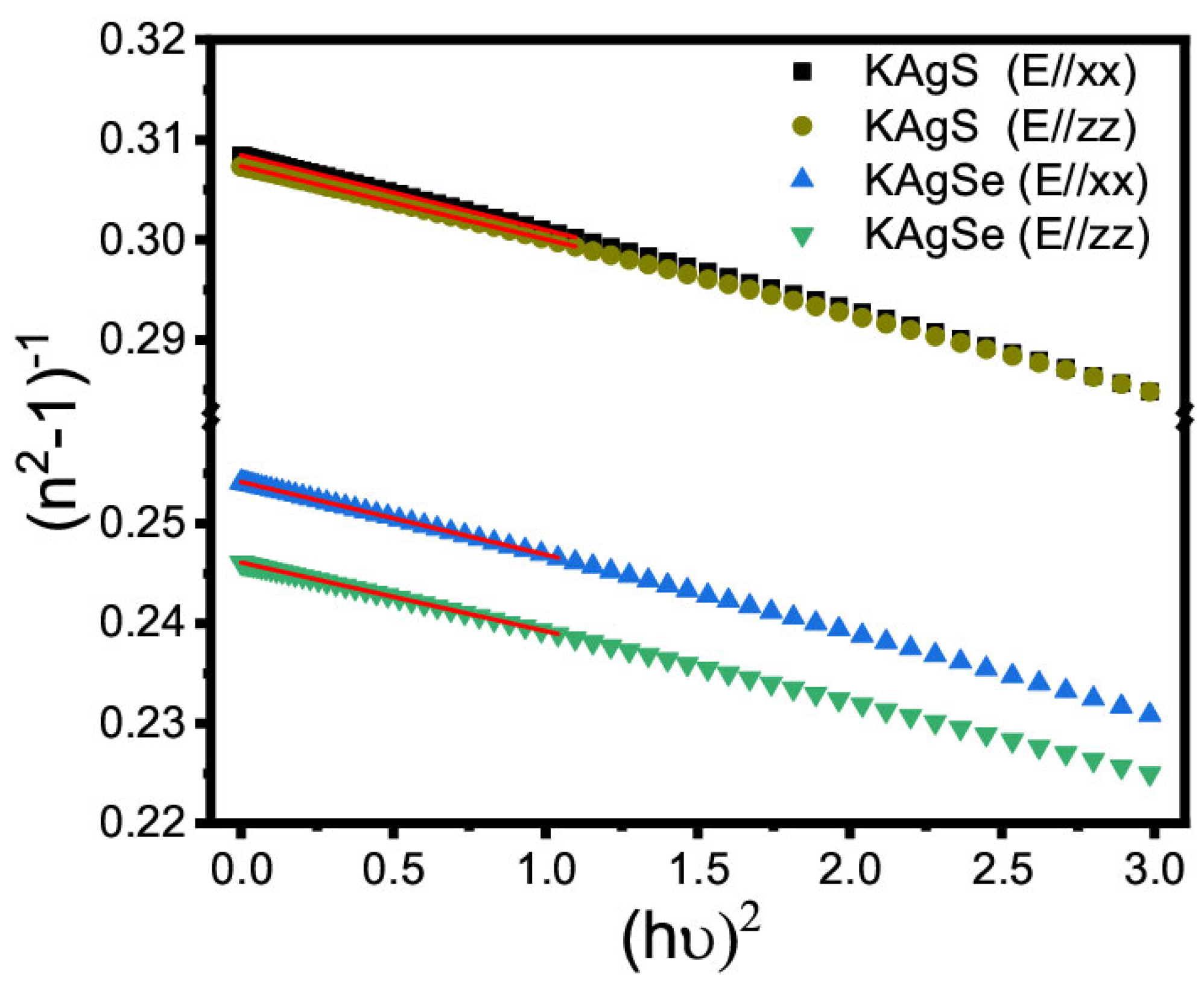
3.4.2. Nonlinear Optical Parameters
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pan, S.; Smit, J.P.; Watkins, B.; Marvel, M.R.; Stern, C.L.; Poeppelmeier, K.R. Synthesis, crystal structure, and nonlinear optical properties of Li6CuB4O10: A congruently melting compound with isolated [CuB4O10] 6-units. J. Am. Chem. Soc. 2006, 128, 11631–11634. [Google Scholar] [CrossRef] [PubMed]
- Choyke, S.J.; Blau, S.M.; Larner, A.A.; Sarjeant, A.N.; Yeon, J.; Halasyamani, P.S.; Norquist, A.J. Noncentrosymmetry in new templated gallium fluorophosphates. Inorg. Chem. 2009, 48, 11277–11282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-L.; Cheng, W.-D.; Zhang, H.; Geng, L.; Lin, C.-S.; He, Z.-Z. A strong second-harmonic generation material Cd4BiO (BO3) 3 originating from 3-chromophore asymmetric structures. J. Am. Chem. Soc. 2010, 132, 1508–1509. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-F.; Hu, C.-L.; Xu, X.; Yang, B.-P.; Mao, J.-G. Explorations of new second-order nonlinear optical materials in the potassium vanadyl iodate system. J. Am. Chem. Soc. 2011, 133, 5561–5572. [Google Scholar] [CrossRef]
- Medhekar, S.; Sarkar, R.K. All-optical passive transistor. Opt. Lett. 2005, 30, 887–889. [Google Scholar] [CrossRef]
- Sarkar, R.K.; Medhekar, S. Passive,“self-trapped family” all-optical half adder using all-optical XOR and AND gates. Czechoslov. J. Phys. 2006, 56, 359–366. [Google Scholar] [CrossRef]
- Atuchin, V.V.; Bazarov, B.G.; Gavrilova, T.A.; Grossman, V.G.; Molokeev, M.S.; Bazarova, Z.G. Preparation and structural properties of nonlinear optical borates K2 (1 − x) Rb2xAl2B2O7, 0 < x< 0.75. J. Alloy. Compd. 2012, 515, 119–122. [Google Scholar]
- Tran, T.T.; Koocher, N.Z.; Rondinelli, J.M.; Halasyamani, P.S. Beryllium-free β-Rb2Al2B2O7 as a possible deep-ultraviolet nonlinear optical material replacement for KBe2BO3F2. Angew. Chem. 2017, 129, 3015–3019. [Google Scholar] [CrossRef]
- Feng, J.; Hu, C.; Xu, X.; Li, B.; Zhang, M.; Mao, J. AgGa2PS6: A New Mid-Infrared Nonlinear Optical Material with a High Laser Damage Threshold and a Large Second Harmonic Generation Response. Chem. Eur. J. 2017, 23, 10978–10982. [Google Scholar] [CrossRef]
- Behera, D.; Sharma, R.; Ullah, H.; Waheed, H.S.; Mukherjee, S.K. Electronic, optical, and thermoelectric investigations of Zintl phase AAg2Se2 (A = Sr, Ba) compounds: A first first-principles approach. J. Solid State Chem. 2022, 312, 123259. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 11002. [Google Scholar] [CrossRef]
- Wang, Q.; Li, J.; Liang, Y.; Nie, Y.; Wang, B. KAgSe: A new two-dimensional efficient photovoltaic material with layer-independent behaviors. ACS Appl. Mater. Interfaces 2018, 10, 41670–41677. [Google Scholar] [CrossRef] [PubMed]
- Savelsberg, G.; Schäfer, H. Beiträge zu den Stabilitätskriterien des PbFCl-typs: Darstellung und Struktur von KAgSe. J. Less Common Met. 1981, 80, P59–P69. [Google Scholar] [CrossRef]
- Ding, L.-J.; Li, G.-G.; Zhang, C.-W.; Li, P.; Wang, P.-J. Novel two-dimensional KAB (A = Cu, Au, B = S, Se) photoelectric materials with Prominent carrier mobility and optical properties. Superlattices Microstruct. 2021, 149, 106773. [Google Scholar] [CrossRef]
- Boualleg, M.; Bennecer, B.; Kalarasse, F. Ab initio predictions of structures and physical properties of the KCuX (X = Se and Te) phases under pressure. Comput. Condens. Matter 2022, 30, e00616. [Google Scholar] [CrossRef]
- Savelsberg, G. Ternäre pnictide und chalkogenide von alkalimetallen und IB-bzw. IIB-elementen/On ternary pnictides and chalkogenides of alkaline metals and IB-resp. II B-elements. Z. Naturforsch. B 1978, 33, 370–373. [Google Scholar] [CrossRef]
- Mahmoud, M.; Rugut, E.K.; Molepo, M.P.; Joubert, D.P. First-principles study of structural stability, electronic properties and lattice thermal conductivity of KAgX (X = S, Se, Te). Eur. Phys. J. B 2019, 92, 1–10. [Google Scholar] [CrossRef]
- Basri, S.; Zulkifli, M.E.; Hazri, N.S.; Kamarudin, S.K. Quantum Behaviour of Mg and Mg-Al-Zn Microstructure. Crystals 2023, 13, 501. [Google Scholar] [CrossRef]
- Mounet, N.; Gibertini, M.; Schwaller, P.; Campi, D.; Merkys, A.; Marrazzo, A.; Sohier, T.; Castelli, I.E.; Cepellotti, A.; Pizzi, G. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 2018, 13, 246–252. [Google Scholar] [CrossRef]
- Xu, W.; Wang, R.; Zheng, B.; Wu, X.; Xu, H. New family of two-dimensional ternary photoelectric materials. ACS Appl. Mater. Interfaces 2019, 11, 14457–14462. [Google Scholar] [CrossRef]
- Zhu, X.-L.; Yang, H.; Zhou, W.-X.; Wang, B.; Xu, N.; Xie, G. KAgX (X = S, Se): High-performance layered thermoelectric materials for medium-temperature applications. ACS Appl. Mater. Interfaces 2020, 12, 36102–36109. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liang, Y.; Yao, H.; Li, J.; Wang, B.; Wang, J. Emerging negative differential resistance effects and novel tunable electronic behaviors of the broken-gap KAgSe/SiC 2 van der Waals heterojunction. J. Mater. Chem. C 2020, 8, 8107–8119. [Google Scholar] [CrossRef]
- Konar, S.; Biswas, A. Soliton-soliton interaction with Kerr law nonlinearity. J. Electromagn. Waves Appl. 2005, 19, 1443–1453. [Google Scholar] [CrossRef]
- Medhekar, S.; Sarkar, R.K.; Paltani, P.P. Coupled spatial-soliton pairs in saturable nonlinear media. Opt. Lett. 2006, 31, 77–79. [Google Scholar] [CrossRef]
- Khalique, C.M.; Biswas, A. Optical solitons with parabolic and dual-power law nonlinearity via Lie symmetry analysis. J. Electromagn. Waves Appl. 2009, 23, 963–973. [Google Scholar] [CrossRef]
- Sarkar, R.K.; Medhekar, S. Spatial soliton pairing of two cylindrical beams in saturable nonlinear media. Prog. Electromagn. Res. M 2009, 9, 53–64. [Google Scholar] [CrossRef]
- Biswas, A.; Yıldırım, Y.; Yaşar, E.; Zhou, Q.; Alshomrani, A.S.; Belic, M. Optical soliton perturbation in parabolic law medium having weak non-local nonlinearity by a couple of strategic integration architectures. Results Phys. 2019, 13, 102334. [Google Scholar] [CrossRef]
- Medhekar, S.; Sarkar, R.K.; Paltani, P.P. Soliton pairing of two coaxially co-propagating mutually incoherent 1-D beams in Kerr type media. Opt. Appl. 2007, 37, 243. [Google Scholar]
- Biswas, A.; Kara, A.H.; Khan, S.; Yildirim, Y.; Mahmood, M.F.; Alshehri, H.M.; Belic, M.R. Conservation laws for cubic–quartic optical solitons with complex Ginzburg–Landau equation having five nonlinear refractive index structures. Adv. Mater. Rapid Commun. 2022, 16, 137–141. [Google Scholar]
- Shwetanshumala, S.; Konar, S. Bright optical spatial solitons in a photorefractive waveguide. Phys. Scr. 2010, 82, 45404. [Google Scholar] [CrossRef]
- Biswas, A.; Sonmezoglu, A.; Ekici, M.; Alzahrani, A.K.; Belic, M.R. Cubic–quartic optical solitons with differential group delay for Kudryashov’s model by extended trial function. J. Commun. Technol. Electron. 2020, 65, 1384–1398. [Google Scholar] [CrossRef]
- Sarkar, R.K.; Medhekar, S. Mutual-focusing of two co-propagating beams and formation of trapped spatial breather pair in saturable nonlinear media. Optik 2010, 121, 339–346. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Madsen, G.; Kvasnicka, D.; Luitz, J. Inst. F. Mater. Chem. TU Vienna. Available online: http://susi.theochem.tuwien.ac.at (accessed on 19 April 2023).
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Density functional theory (DFT). Phys. Rev 1964, 136, B864. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Sorantin, P.; Trickey, S.B. Full-potential, linearized augmented plane wave programs for crystalline systems. Comput. Phys. Commun. 1990, 59, 399–415. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Koller, D.; Tran, F.; Blaha, P. Improving the modified Becke-Johnson exchange potential. Phys. Rev. B 2012, 85, 155109. [Google Scholar] [CrossRef]
- Behera, D.; Manzoor, M.; Iqbal, M.W.; Lakra, S.; Mukherjee, S.K. Revealing Excellent Electronic, Optical, and Thermoelectric Behavior of EU Based Euag2y2 (Y = S/Se): For Solar Cell Applications. Comput. Condens. Matter 2022, 32, e00723. [Google Scholar] [CrossRef]
- Behera, D.; Mukherjee, S.K. Optoelectronics and Transport Phenomena in Rb2InBiX6 (X = Cl, Br) Compounds for Renewable Energy Applications: A DFT Insight. Chemistry 2022, 4, 1044–1059. [Google Scholar] [CrossRef]
- Behera, D.; Mukherjee, S.K. Theoretical Investigation of the Lead-free K2InBiX6 (X = Cl, Br) Double Perovskite Compounds Using First Principle Calculation. JETP Lett. 2022, 116, 1–10. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Luaña, V. Gibbs2: A new version of the quasi-harmonic model code. I. Robust treatment of the static data. Comput. Phys. Commun. 2011, 182, 1708–1720. [Google Scholar] [CrossRef]
- Hafner, J.; Kresse, G. The vienna ab-initio simulation program VASP: An efficient and versatile tool for studying the structural, dynamic, and electronic properties of materials. In Properties of Complex Inorganic Solids; Springer: Berlin/Heidelberg, Germany, 1997; pp. 69–82. [Google Scholar]
- Saal, J.E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials design and discovery with high-throughput density functional theory: The open quantum materials database (OQMD). JOM 2013, 65, 1501–1509. [Google Scholar] [CrossRef]
- Manzoor, M.; Behera, D.; Sharma, R.; Iqbal, M.W.; Mukherjee, S.K.; Khenata, R.; Alarfaji, S.S.; Alzahrani, H.A. Investigation of the structural, mechanical, optoelectronic and, thermoelectric characteristics of cubic GeTiO3: An ab initio study. Mater. Today Commun. 2023, 34, 105053. [Google Scholar] [CrossRef]
- Behera, D.; Manzoor, M.; Maharana, M.; Iqbal, M.W.; Zahid, T.; Lakra, S.; Mukherjee, S.K.; Alarfaji, S.S. Structural, electronic, optical, and thermoelectric response of zintl phase AAg2S2 (A= Sr/Ba) compounds for renewable energy applications. Phys. B Condens. Matter 2023, 649, 414446. [Google Scholar] [CrossRef]
- Manzoor, M.; Chowdhury, S.; Sharma, R.; Iqbal, M.W.; Mukherjee, S.K.; Alarfaji, S.S.; Alzahrani, H.A. Insight on the lattice dynamics, thermodynamic and thermoelectric properties of CdYF3 perovskite: A DFT study. Comput. Theor. Chem. 2022, 113928. [Google Scholar] [CrossRef]
- Abraham, J.A.; Behera, D.; Kumari, K.; Srivastava, A.; Sharma, R.; Mukherjee, S.K. A comprehensive DFT analysis on structural, electronic, optical, thermoelectric, SLME properties of new Double Perovskite Oxide Pb2ScBiO6. Chem. Phys. Lett. 2022, 806, 139987. [Google Scholar] [CrossRef]
- Behera, D.; Mukherjee, S.K. Structural, elastic, electronic and thermoelectric properties of K2GeBr6: A first principle approach. Mater. Today Proc. 2023. [Google Scholar] [CrossRef]
- Koller, D.; Tran, F.; Blaha, P. Merits and limits of the modified Becke-Johnson exchange potential. Phys. Rev. B 2011, 83, 195134. [Google Scholar] [CrossRef]
- Rubel, O.; Tran, F.; Rocquefelte, X.; Blaha, P. Perturbation approach to ab initio effective mass calculations. Comput. Phys. Commun. 2021, 261, 107648. [Google Scholar] [CrossRef]
- Behera, D.; Abraham, J.A.; Sharma, R.; Mukerjee, S.K.; Jain, E. First Principles Study of New d0 Half-Metallic Ferromagnetism in CsBaC Ternary Half-Heusler Alloy. J. Supercond. Nov. Magn. 2022, 35, 3431–3437. [Google Scholar] [CrossRef]
- Behera, D.; Dixit, A.; Nahak, B.; Srivastava, A.; Dubey, S.; Sharma, R.; Mishra, A.K.; Mukherjee, S.K. Structural, electronic, elastic, vibrational and thermodynamic properties of antiperovskites Mg3NX (X = Ge, Sn): A DFT study. Phys. Lett. A 2022, 453, 128478. [Google Scholar] [CrossRef]
- Fitzgerel, R.K.; Verhoek, F.H. The law of Dulong and Petit 1960. J. Chem. Educ. 1960, 37, 545. [Google Scholar] [CrossRef]
- Inaba, H.; Yamamoto, T. Debye temperature of materials. Netsu Sokutei 1983, 10, 132–145. [Google Scholar]
- Behera, D.; Dixit, A.; Kumari, K.; Srivastava, A.; Sharma, R.; Mukherjee, S.K.; Khenata, R.; Boumaza, A.; Bin-Omran, S. Structural, elastic, mechanical, and thermodynamic characteristic of NaReO3 and KReO3 perovskite oxides from first principles study. Eur. Phys. J. Plus 2022, 137, 1345. [Google Scholar] [CrossRef]
- Behera, D.; Manzoor, M.; Sharma, R.; Iqbal, M.W.; Mukherjee, S.K. First principle insight on structural, opto-electronic and transport properties of novel zintl-phase AMg2Bi2 (A = Sr, Ba). J. Solid State Chem. 2023, 320, 123860. [Google Scholar] [CrossRef]
- Ouerghui, W.; Alkhalifah, M.S. Density functional investigation of structural, electronic, optical and thermodynamic properties of Zn1− xBexO semiconductor. Appl. Phys. A 2019, 125, 1–12. [Google Scholar] [CrossRef]
- Ben Abdallah, H.; Ouerghui, W. Hybrid functional calculations of electro-optical properties of novel Ga1−xInxTe ternary chalcogenides. Appl. Phys. A 2020, 126, 1–12. [Google Scholar] [CrossRef]
- Behera, D.; Manzoor, M.; Mukherjee, S.K. Incorporation of Te in enhancing thermoelectric response of AeAg2SeTe (Ae = Sr, Ba) compounds: A DFT insight. Comput. Condens. Matter 2022, 33, e00757. [Google Scholar] [CrossRef]
- Joshi, H.; Rai, D.P.; Hnamte, L.; Laref, A.; Thapa, R.K. A theoretical analysis of elastic and optical properties of half Heusler MCoSb (M = Ti, Zr and Hf). Heliyon 2019, 5, e01155. [Google Scholar] [CrossRef]
- Manzoor, M.; Bahera, D.; Sharma, R.; Tufail, F.; Iqbal, M.W.; Mukerjee, S.K. Investigated the structural, optoelectronic, mechanical, and thermoelectric properties of Sr2BTaO6 (B = Sb, Bi) for solar cell applications. Int. J. Energy Res. 2022, 46, 23698–23714. [Google Scholar] [CrossRef]
- Ouerghui, W.; Alkhalifah, M.S.; Abdallah, H. Ben DFT calculations on ZnO1− x compounds for optoelectronic applications. J. Comput. Electron. 2021, 20, 467–479. [Google Scholar] [CrossRef]
- Alkhalifah, M.S.; Ouerghui, W. DFT calculations of optoelectronic properties of cubic $$\left ({{\text {In}}_{1-x}{\text {Al}}_{x}}\right)_{2}{\text {O}}_{3} $$ In 1-x Al x 2 O 3 alloys. J. Comput. Electron. 2021, 20, 1234–1247. [Google Scholar] [CrossRef]
- Babushkina, E.A.; Belokopytova, L.V.; Grachev, A.M.; Meko, D.M.; Vaganov, E.A. Variation of the hydrological regime of Bele-Shira closed basin in Southern Siberia and its reflection in the radial growth of Larix sibirica. Reg. Environ. Chang. 2017, 17, 1725–1737. [Google Scholar] [CrossRef]
- Ouerghui, W.; Gassoumi, M.; Beji, L.; Maaref, M.A. Optical properties of quaternary GaMnAsP thin layer grown by molecular beam epitaxy. Phys. E Low-Dimens. Syst. Nanostructures 2021, 131, 114733. [Google Scholar] [CrossRef]
- Msalmi, R.; Elleuch, S.; Hamdi, B.; Zouari, R.; Naïli, H. Synthesis, DFT calculations, intermolecular interactions and third order nonlinear optical properties of new organoammonium tetrabromocadmate (II):(C5H6N2Cl) 2 [CdBr4]·H2O. J. Mol. Struct. 2020, 1222, 128853. [Google Scholar] [CrossRef]
- Sharda, S.; Sharma, N.; Sharma, P.; Sharma, V. New quaternary Sb-Se-Ge-In chalcogenide glasses: Linear and nonlinear optical properties. J. Electron. Mater. 2013, 42, 3367–3372. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a = b | c | c/a | B | B’ | zK | wCh | Ef | ||
|---|---|---|---|---|---|---|---|---|---|
| KAgS | This work | 4.39 | 7.56 | 1.72 | 39.14 | 4.41 | 0.657 | 0.216 | −0.71 |
| Expt. | |||||||||
| Other works | 4.391 [44] | 7.556 [44] | |||||||
| 4.42 [17] | 7.53 [17] | 1.70 [17] | 29.96 [17] | 6.61 [17] | |||||
| 4.32 [17] | 7.36 [17] | 34.79 [17] | 7.18 [17] | ||||||
| KAgSe | This work | 4.51 | 7.72 | 1.71 | 35.37 | 5.00 | 0.654 | 0.231 | −0.61 |
| Expt. [10] | 4.52 | 7.59 | 1.68 | 0.6579 | 0.2156 | ||||
| Other works | 4.29 [44] | 7.275 [44] | |||||||
| 4.56 [11] | 7.77 [11] | 1.70 [11] | 25.87 [17] | 6.47 [17] | |||||
| 4.57 [17] | 7.79 [17] | 30.03 [17] | 6.99 [17] | ||||||
| 4.47 [17] | 7.61 [17] |
| Compound | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (E//x) | (E//z) | (E//x) | (E//z) | (E//x) | (E//z) | (E//x) | (E//z) | (E//x) | (E//z) | |
| KAgS | 4.24 | 4.25 | 2.05 | 2.06 | 6.28 | 6.40 | 20.36 | 20.82 | 128.02 | 133.32 |
| KAgSe | 4.93 | 5.06 | 2.22 | 2.24 | 5.75 | 5.98 | 22.64 | 24.03 | 130.36 | 143.72 |
| Compound | ||||
|---|---|---|---|---|
| (E//x) | (E//z) | (E//x) | (E//z) | |
| KAgS | 1.37 | 1.39 | 7.51 | 7.62 |
| KAgSe | 2.75 | 3.09 | 16.2 | 18.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seddik, T.; Behera, D.; Batouche, M.; Ouerghui, W.; Abdallah, H.B.; Sarkar, R.K.; Salah, M.M.; Shaker, A.; Mukherjee, S.K. Electronic Properties, Linear and Nonlinear Performance of KAgCh (Ch = S, Se) Compounds: A First-Principles Study. Crystals 2023, 13, 726. https://doi.org/10.3390/cryst13050726
Seddik T, Behera D, Batouche M, Ouerghui W, Abdallah HB, Sarkar RK, Salah MM, Shaker A, Mukherjee SK. Electronic Properties, Linear and Nonlinear Performance of KAgCh (Ch = S, Se) Compounds: A First-Principles Study. Crystals. 2023; 13(5):726. https://doi.org/10.3390/cryst13050726
Chicago/Turabian StyleSeddik, Taieb, Debidatta Behera, Mohammed Batouche, Walid Ouerghui, Houda Ben Abdallah, Ram Krishna Sarkar, Mostafa M. Salah, Ahmed Shaker, and Sanat Kumar Mukherjee. 2023. "Electronic Properties, Linear and Nonlinear Performance of KAgCh (Ch = S, Se) Compounds: A First-Principles Study" Crystals 13, no. 5: 726. https://doi.org/10.3390/cryst13050726
APA StyleSeddik, T., Behera, D., Batouche, M., Ouerghui, W., Abdallah, H. B., Sarkar, R. K., Salah, M. M., Shaker, A., & Mukherjee, S. K. (2023). Electronic Properties, Linear and Nonlinear Performance of KAgCh (Ch = S, Se) Compounds: A First-Principles Study. Crystals, 13(5), 726. https://doi.org/10.3390/cryst13050726