
The Crystal Structure of Pb10(PO4)6O Revisited: The Evidence of Superstructure


Abstract
:1. Introduction
2. Background Information
3. Materials and Methods
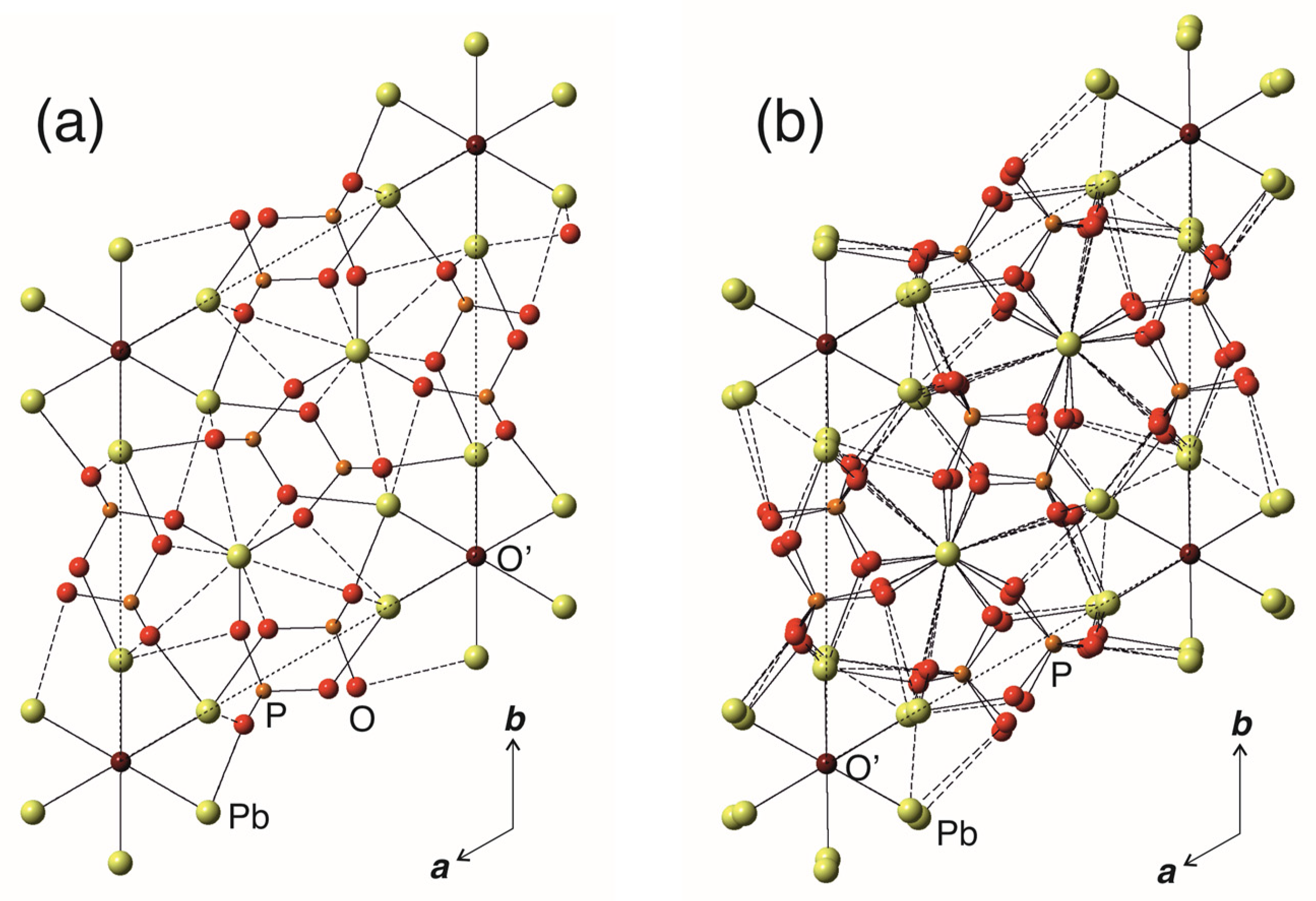
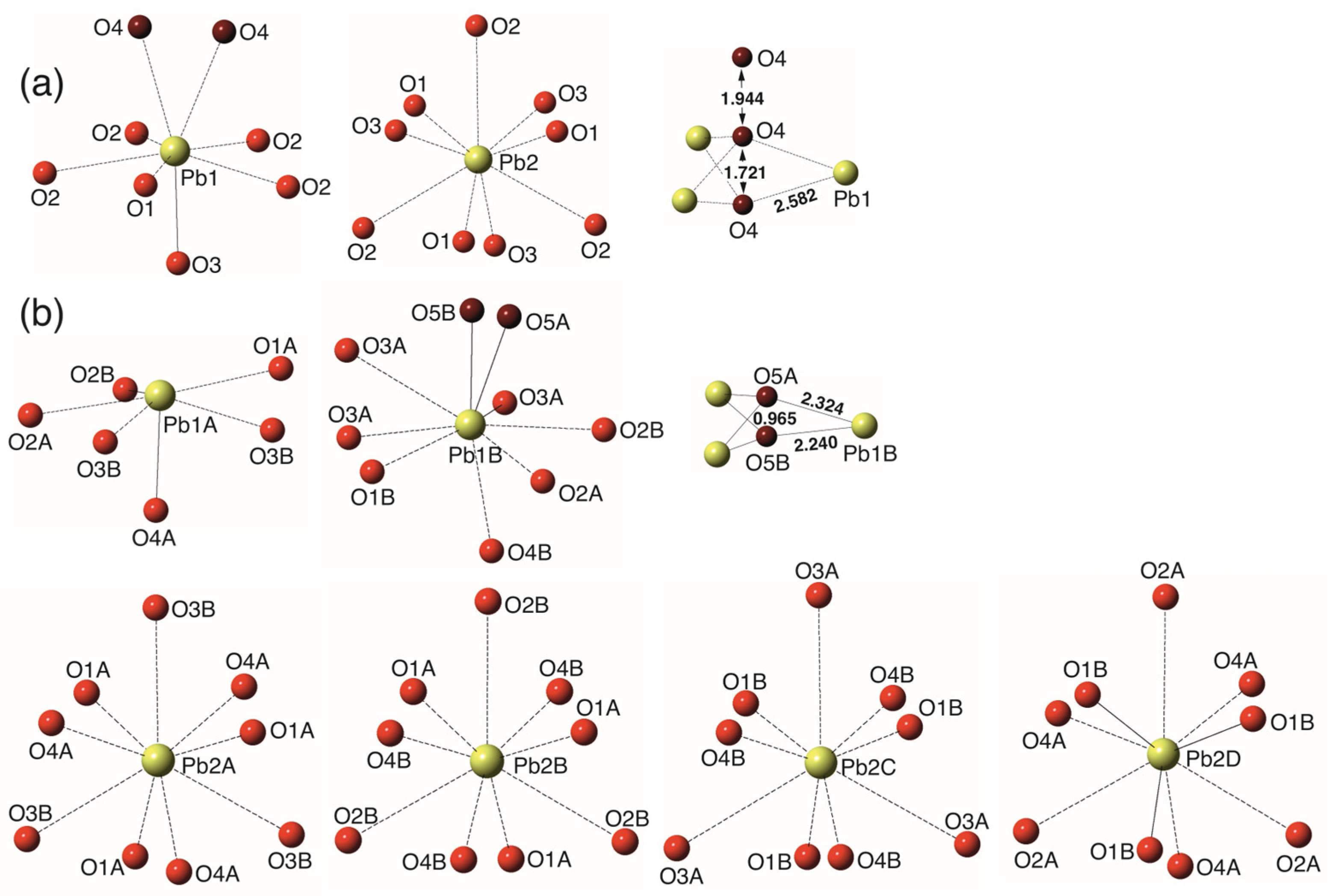
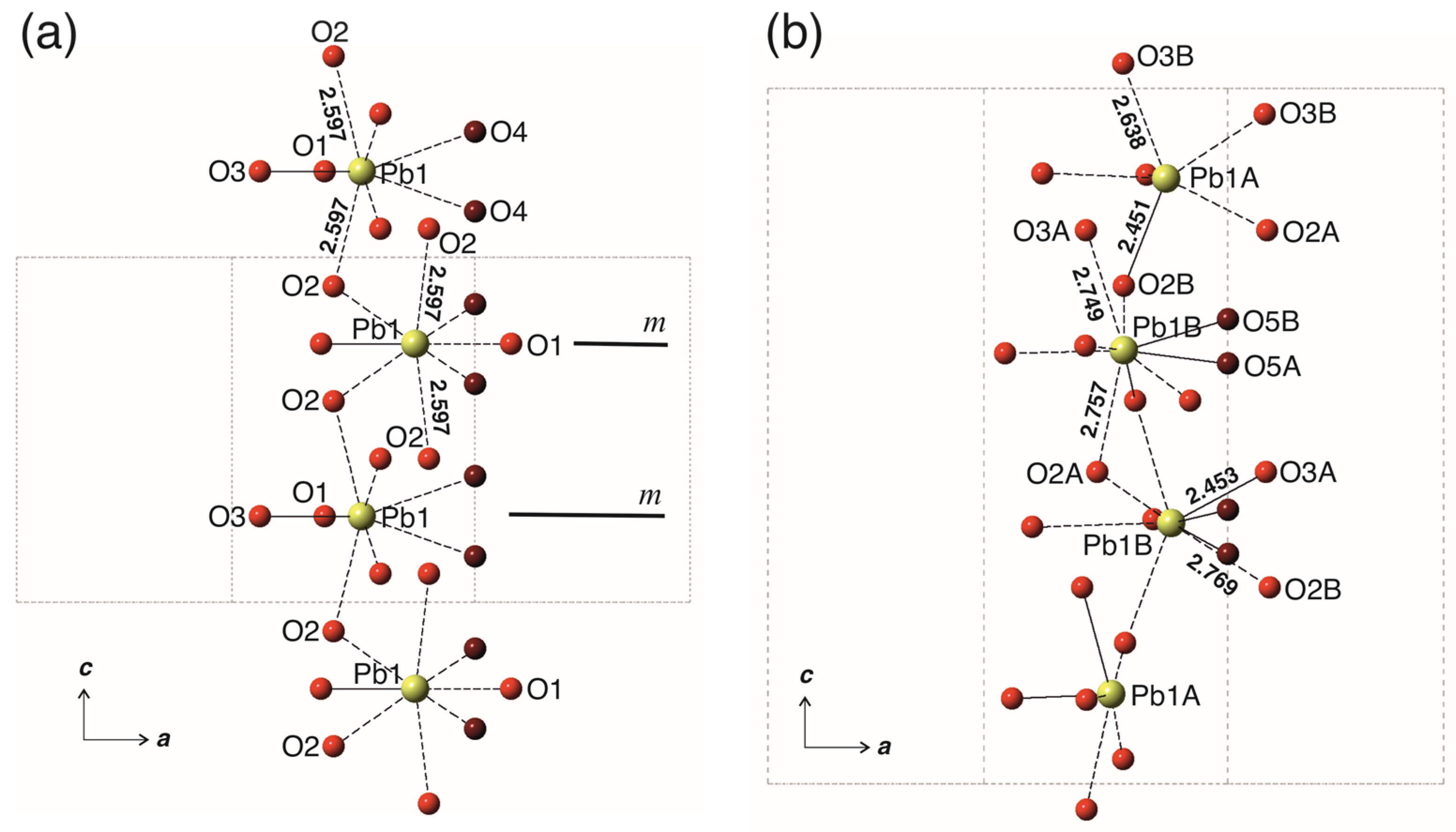
4. Results
5. Discussion
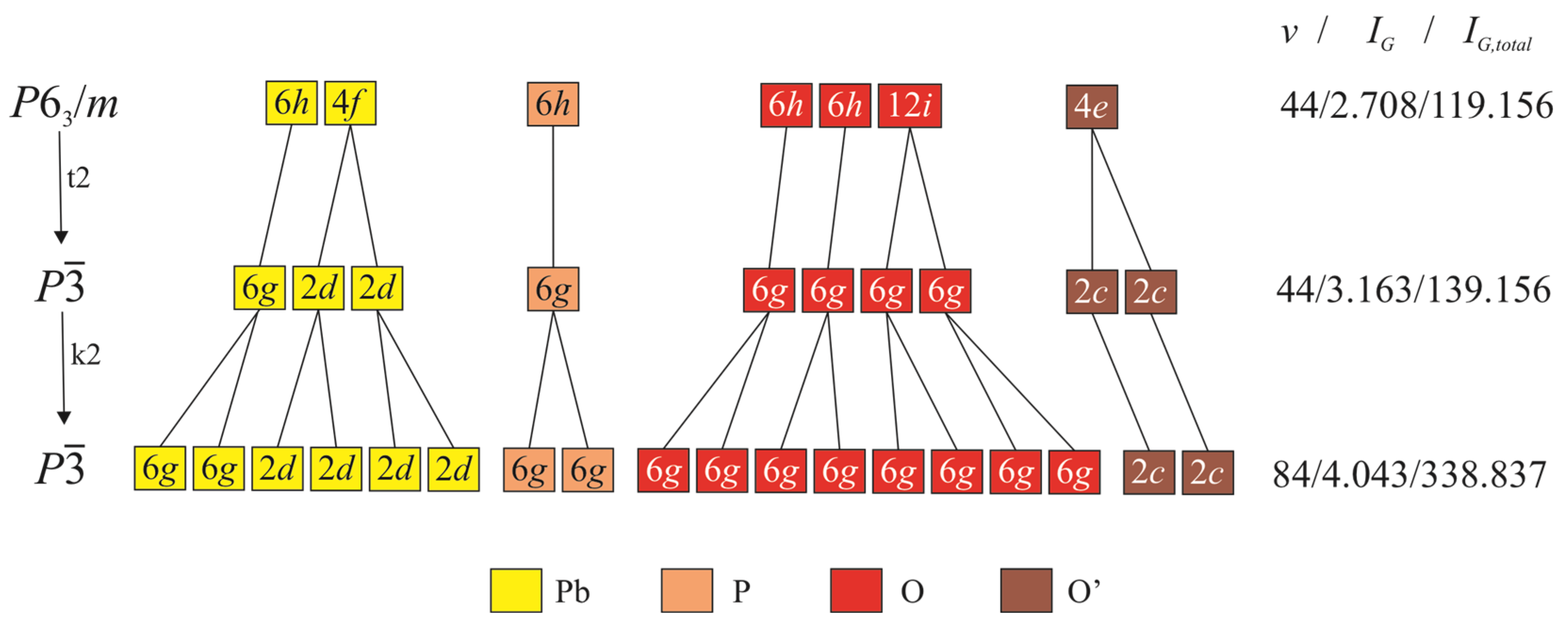
5.1. Symmetry Considerations
5.2. The Relations between KB and WE
5.3. Structural Complexity
5.4. On the Mechanism of the Pb-Cu Substitution in ‘Oxypyromorphite’
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, S.; Kim, J.; Im, S.; An, S.M.; Kwon, Y.-W.; Ho, A. Consideration for the Development of Room-Temperature Ambient-Pressure Superconductor (LK-99). J. Korean Crystal Growth Crystal Technol. 2023, 33, 61–70. [Google Scholar]
- Lee, S.; Kim, J.; Kim, H.-T.; Im, S.; An, S.; Auh, K.H. Superconductor Pb10−xCux(PO4)6O Showing Levitation at Room Temperature and Atmospheric Pressure and Mechanism. arXiv 2023, arXiv:2307.12037v2. [Google Scholar]
- Lee, S.; Kim, J.-H.; Kwon, Y.-W. The First Room-Temperature Ambient-Pressure Superconductor. arXiv 2023, arXiv:2307.12008. [Google Scholar]
- Hou, Q.; Wei, W.; Zhou, X.; Sun, Y.; Shi, Z. Observation of Zero Resistance above 100 K in Pb10−xCux(PO4)6O. arXiv 2023, arXiv:2308.01192v1. [Google Scholar]
- Kurleto, R.; Lany, S.; Pashov, D.; Acharya, S.; van Schilfgaarde, M.; Dessau, D.S. Pb-Apatite Framework as a Generator of Novel Flat-Band CuO Based Physics, Including Possible Room Temperature Superconductivity. arXiv 2023, arXiv:2308.00698. [Google Scholar]
- Liu, L.; Meng, Z.; Wang, X.; Chen, H.; Duan, Z.; Zhou, X.; Yan, H.; Qin, P.; Liu, Z. Semiconducting Transport in Pb10−xCux(PO4)6O Sintered from Pb₂SO₅ and Cu₃P. arXiv 2023, arXiv:2307.16802. [Google Scholar]
- Abramian, P.; Kuzanyan, A.; Nikoghosyan, V.; Teknowijoyo, S.; Gulian, A. Some Remarks on Possible Superconductivity of Composition Pb₉CuP6O25. arXiv 2023, arXiv:2308.01723. [Google Scholar]
- Wu, H.; Yang, L.; Xiao, B.; Chang, H. Successful Growth and Room Temperature Ambient-Pressure Magnetic Levitation of LK-99. arXiv 2023, arXiv:2308.01516. [Google Scholar]
- Si, L.; Held, K. Electronic Structure of the Putative Room-Temperature Superconductor Pb₉Cu(PO4)6O. arXiv 2023, arXiv:2308.00676. [Google Scholar]
- Lai, J.; Li, J.; Liu, P.; Sun, Y.; Chen, X.-Q. First-Principles Study on the Electronic Structure of Pb10−xCux(PO4)6O (x = 0, 1). J. Mater. Sci. Technol. 2023, 171, 66–70. [Google Scholar] [CrossRef]
- Tavakol, O.; Scaffidi, T. Minimal Model for the Flat Bands in Copper-Substituted Lead Phosphate Apatite. arXiv 2023, arXiv:2308.01315. [Google Scholar]
- Griffin, S.M. Origin of Correlated Isolated Flat Bands in Copper-Substituted Lead Phosphate Apatite. arXiv 2023, arXiv:2307.16892. [Google Scholar]
- Cabezas-Escares, J.; Barrera, N.F.; Cardenas, C.; Munoz, F. Theoretical Insight on the LK-99 Material. arXiv 2023, arXiv:2308.01135. [Google Scholar]
- Zhou, B.T.; Franz, M. Cu-Substituted Lead Phosphate Apatite as an Inversion-Asymmetric Weyl Semimetal. arXiv 2023, arXiv:2308.07408. [Google Scholar]
- Hou, Q.; Wei, W.; Zhou, X.; Wang, X.; Sun, Y.; Shi, Z. Current Percolation Model for the Special Resistivity Behavior Observed in Cu-Doped Apatite. arXiv 2023, arXiv:2308.05778. [Google Scholar]
- Liu, R.; Guo, T.; Lu, J.; Ren, J.; Ma, T. Different Phase Leads to Different Transport Behavior in Pb₉Cu(PO4)6O Compounds. arXiv 2023, arXiv:2308.08454. [Google Scholar]
- Korotin, D.M.; Novoselov, D.Y.; Shorikov, A.O.; Anisimov, V.I.; Oganov, A.R. Electronic Correlations in Promising Room-Temperature Superconductor Pb₉Cu(PO4)6O: A DFT+DMFT Study. arXiv 2023, arXiv:2308.04301. [Google Scholar]
- Panas, I. Entertaining the Possibility of RT Superconductivity in LK-99. arXiv 2023, arXiv:2308.06684. [Google Scholar]
- Zhang, Y.; Liu, C.; Zhu, X.; Wen, H.-H. Ferromagnetism and Insulating Behavior with a Logarithmic Temperature Dependence of Resistivity in Pb10−xCux(PO4)6O. arXiv 2023, arXiv:2308.05786. [Google Scholar]
- Liu, L.; Ren, X.; Hu, J. Metallic States in Pb10(PO4)6O Induced by the Cu/O-Insertions and Carrier Doping. arXiv 2023, arXiv:2308.07345. [Google Scholar]
- Witt, N.; Si, L.; Tomczak, J.M.; Held, K.; Wehling, T. No Superconductivity in Pb₉Cu₁(PO4)6O Found in Orbital and Spin Fluctuation Exchange Calculations. arXiv 2023, arXiv:2308.07261. [Google Scholar]
- Singh, H.; Gautam, A.; Singh, M.; Saha, P.; Kumar, P.; Das, P.; Lamba, M.; Yadav, K.; Mishra, P.K.; Patnaik, S.; et al. On the Experimental Evidence for Possible Superconductivity in LK99. arXiv 2023, arXiv:2308.06589. [Google Scholar]
- Thakur, G.S.; Schulze, M.; Ruck, M. On the Synthesis Methodologies to Prepare Pb₉Cu(PO4)6O—Phase, Composition, Magnetic Analysis and Absence of Superconductivity. arXiv 2023, arXiv:2308.05776. [Google Scholar]
- Jiang, Y.; Lee, S.B.; Herzog-Arbeitman, J.; Yu, J.; Feng, X.; Hu, H.; Călugăru, D.; Brodale, P.S.; Gormley, E.L.; Vergniory, M.G.; et al. Pb₉Cu(PO4)6(OH)₂: Phonon Bands, Localized Flat Band Magnetism, Models, and Chemical Analysis. arXiv 2023, arXiv:2308.05143. [Google Scholar]
- Shen, J.; Gaines, D.; Shahabfar, S.; Li, Z.; Kang, D.; Griesemer, S.; Salgado-Casanova, A.; Liu, T.; Chou, C.-T.; Xia, Y.; et al. Phase Stability of Lead Phosphate Apatite Pb10−xCux(PO4)6O, Pb10-xCux(PO4)6(OH)₂, and Pb₈Cu₂(PO4)6. arXiv 2023, arXiv:2308.07941. [Google Scholar]
- Liu, C.; Cheng, W.; Zhang, X.; Xu, J.; Li, J.; Shi, Q.; Yuan, C.; Xu, L.; Zhou, H.; Zhu, S.; et al. Phases and Magnetism at the Microscale in Compounds Containing Nominal Pb10−xCux(PO4)6O. arXiv 2023, arXiv:2308.07800. [Google Scholar] [CrossRef]
- Hlinka, J. Possible Ferroic Properties of Copper-Substituted Lead Phosphate Apatite. arXiv 2023, arXiv:2308.03691. [Google Scholar]
- Puphal, P.; Akbar, M.Y.P.; Hepting, M.; Goering, E.; Isobe, M.; Nugroho, A.A.; Keimer, B. Single Crystal Synthesis, Structure, and Magnetism of Pb10−xCu_x(PO4)6O. arXiv 2023, arXiv:2308.06256. [Google Scholar]
- Chen, N.; Liu, Y.; Li, Y. Statistical Material Law Support for Room Temperature Superconductivity in the Lead Apatite System. arXiv 2023, arXiv:2308.06349. [Google Scholar]
- Zhang, J.; Li, H.; Yan, M.; Gao, M.; Ma, F.; Yan, X.-W.; Xie, Z.Y. Structural, Electronic, Magnetic Properties of Cu-Doped Lead-Apatite Pb10−xCu_x(PO4)6O. arXiv 2023, arXiv:2308.04344. [Google Scholar]
- Liu, J.; Yu, T.; Li, J.; Wang, J.; Lai, J.; Sun, Y.; Chen, X.-Q.; Liu, P. Symmetry Breaking Induced Insulating Electronic State in Pb9Cu(PO4)6O. Available online: https://www.researchgate.net/publication/373216676 (accessed on 7 August 2023).
- Timokhin, I.; Chen, C.; Wang, Z.; Yang, Q.; Mishchenko, A. Synthesis and Characterisation of LK-99. arXiv 2023, arXiv:2308.03823. [Google Scholar]
- Vayssilov, G.; Petkov, P.; Aleksandrov, H. Alternative Concept of One-Dimensional Superconductivity—Key Role of Defects Revealed by Quantum Chemical Calculations of Lead Apatite. ChemRxiv. Cambridge: Cambridge Open Engage. Mater. Chem. 2023. [Google Scholar] [CrossRef]
- Krivovichev, S.V.; Burns, P.C. Crystal Chemistry of Lead Oxide Phosphates: Crystal Structures of Pb4O(PO4)2, Pb8O5(PO4)2 and Pb10(PO4)6O. Z. Kristallogr. 2003, 218, 357–365. [Google Scholar] [CrossRef]
- Brückner, S.; Lusvardi, G.; Menabue, L.; Saladini, M. Crystal Structure of Lead Hydroxyapatite from Powder X-ray Diffraction Data. Inorganica Chim. Acta 1995, 236, 209–212. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hunter, B.A.; Fenton, R.R.; Kennedy, B.J. Neutron Powder Diffraction Study of Lead Hydroxyapatite. Aust. J. Chem. 1998, 50, 1061–1066. [Google Scholar] [CrossRef]
- White, T.J.; ZhiLi, D. Structural Derivation and Crystal Chemistry of Apatites. Acta Crystallogr. 2003, B59, 1–16. [Google Scholar] [CrossRef]
- Rooksby, H.P. Identification by X-Ray Diffraction of Crystalline Inclusions in Glass. Analyst 1952, 77, 759–765. [Google Scholar] [CrossRef]
- Merker, L.; Wondratschek, H. Der Oxypromorphit Pb10(PO4)6O und der Ausschnitt Pb4P2O9—Pb3(PO4)2 des Systems PbO—P2O5. Z. Anorg. Allg. Chem. 1960, 306, 25–29. [Google Scholar] [CrossRef]
- Merker, L.; Engel, G.; Wondratschek, H.; Ito, J. Lead Ions and Empty Halide Sites in Apatites. Amer. Miner. 1970, 55, 1435–1437. [Google Scholar]
- Ito, J. Silicate Apatites and Oxyapatites. Amer. Miner. 1968, 53, 890–907. [Google Scholar]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Krivovichev, S.V.; Brown, I.D. Are the Compressive Effects of Encapsulation an Artifact of the Bond Valence Parameters? Z. Kristallogr. 2001, 216, 245–247. [Google Scholar] [CrossRef]
- Gagné, O.C.; Hawthorne, F.C. Comprehensive Derivation of Bond-Valence Parameters for Ion Pairs Involving Oxygen. Acta Crystallogr. 2015, B71, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Müller, U. Symmetry Relationships between Crystal Structures: Applications of Crystallographic Group Theory in Crystal Chemistry; Oxford University Press: Oxford, UK, 2013; ISBN 978-0-19-966995-0. [Google Scholar]
- Siidra, O.I.; Krivovichev, S.V.; Filatov, S.K. Minerals and Synthetic Pb(II) Compounds with Oxocentered Tetrahedra: Review and Classification. Z. Kristallogr. 2008, 223, 114–125. [Google Scholar] [CrossRef]
- Barinova, A.; Bonin, M.; Pushcharovskii, D.; Rastsvetaeva, R.; Schenk, K.; Dimitrova, O. Crystal Structure of Synthetic Hydroxylpyromorphite Pb5(PO4)3(OH). Crystallogr. Rep. 1998, 43, 189–192. [Google Scholar]
- Ben Moussa, S.; Bachoua, H.; Brigui, N.; Badraoui, B. Synthèse, caractérisation et étude structurale des hydroxyapatites à base de plomb Pb10−xMx(PO4)6(OH)2 (M = Zn, Cu). J. Mater. Environ. Sci. 2016, 7, 4754–4766. [Google Scholar]
- Olds, T.A.; Kampf, A.R.; Rakovan, J.F.; Burns, P.C.; Mills, O.P.; Laughlin-Yurs, C. Hydroxylpyromorphite, a Mineral Important to Lead Remediation: Modern Description and Characterization. Amer. Miner. 2021, 106, 922–929. [Google Scholar] [CrossRef]
- Merker, L.; Wondratschek, H. Neue Verbindungen mit apatitartiger Struktur II. Die Gruppe der Alkali-Blei-Verbindungen. Z. Kristallogr. 1957, 109, 110–114. [Google Scholar] [CrossRef]
- Wondratschek, H. Untersuchungen zur Kristallchemie der Blei-Apatite. Neues Jahrb. Miner. Abh. 1963, 99, 113–160, (Habilitation thesis). [Google Scholar]
- Engel, G. Untersuchungen an verschiedenen aus wäßriger Lösung gefällten Bleiphosphaten mit Apatitstruktur. J. Solid State Chem. 1973, 6, 293–298. [Google Scholar] [CrossRef]
- Engel, G. Infrarotspektroskopische und röntgenographische Untersuchungen von Bleihydroxylapatit, Bleioxyapatit und Bleialkaliapatiten. J. Solid State Chem. 1973, 6, 286–292. [Google Scholar] [CrossRef]
- Krivovichev, S.V. Structural Complexity of Minerals: Information Storage and Processing in the Mineral World. Miner. Mag. 2013, 77, 275–326. [Google Scholar] [CrossRef]
- Krivovichev, S.V.; Krivovichev, V.G.; Hazen, R.M.; Aksenov, S.M.; Avdontceva, M.S.; Banaru, A.M.; Gorelova, L.A.; Ismagilova, R.M.; Kornyakov, I.V.; Kuporev, I.V.; et al. Structural and Chemical Complexity of Minerals: An Update. Miner. Mag. 2022, 86, 183–204. [Google Scholar] [CrossRef]
- Liu, R.S.; Hu, S.F.; Chen, D.H.; Shy, D.S.; Jefferson, D.A. Crystal Structure of the (Pb,Hg)Sr2(Ca,Y)Cu2O7-δ Superconductor. Phys. C Supercond. 1994, 222, 13–18. [Google Scholar] [CrossRef]
- Wada, T.; Ichinose, A.; Izumi, F.; Nara, A.; Yamauchi, H.; Hajime-Asano; Tanaka, S. Neutron Powder Diffraction Study of the Pb-Based Copper Oxide Containing Thick Fluorite Blocks: (Pb,Cu)Sr2(Ho,Ce)3Cu2O11+z. Phys. C Supercond. 1991, 179, 455–460. [Google Scholar] [CrossRef]
- Ishigaki, T.; Tokiwa-Yamamoto, A.; Izumi, F.; Kamiyama, T.; Asano, H.; Syono, Y. Structural Changes of PbBaSr(Y0.8Ca0.2)Cu3O7+z on Oxygen Introduction into Block Layers. Phys. C Supercond. 1994, 231, 357–366. [Google Scholar] [CrossRef]
- Jahn, H.A.; Teller, E. Stability of polyatomic molecules in degenerate electronic states. Proc. R. Soc. Ser. A 1937, 161, 220–235. [Google Scholar]
- Hathaway, B.J. A new look at the stereochemistry and electronic properties of complexes of the copper(II) ion. In Complex Chemistry. Structure and Bonding; Emsley, J., Ernst, R.D., Hathaway, B.J., Warren, K.D., Eds.; Springer: Berlin/Heidelberg, Germany, 1984; Volume 57, pp. 55–118. [Google Scholar]
- Burns, P.C.; Hawthorne, F.C. Static and dynamic Jahn-Teller effects in Cu2+ oxysalt minerals. Can. Mineral. 1996, 34, 1089–1105. [Google Scholar]
- Shuvalov, R.R.; Vergasova, L.P.; Semenova, T.F.; Filatov, S.K.; Krivovichev, S.V.; Siidra, O.I.; Rudashevsky, N.S. Prewittite, KPb1.5Cu6Zn(SeO3)2O2Cl10, a New Mineral from Tolbachik Fumaroles, Kamchatka Peninsula, Russia: Description and Crystal Structure. Amer. Miner. 2013, 98, 463–469. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
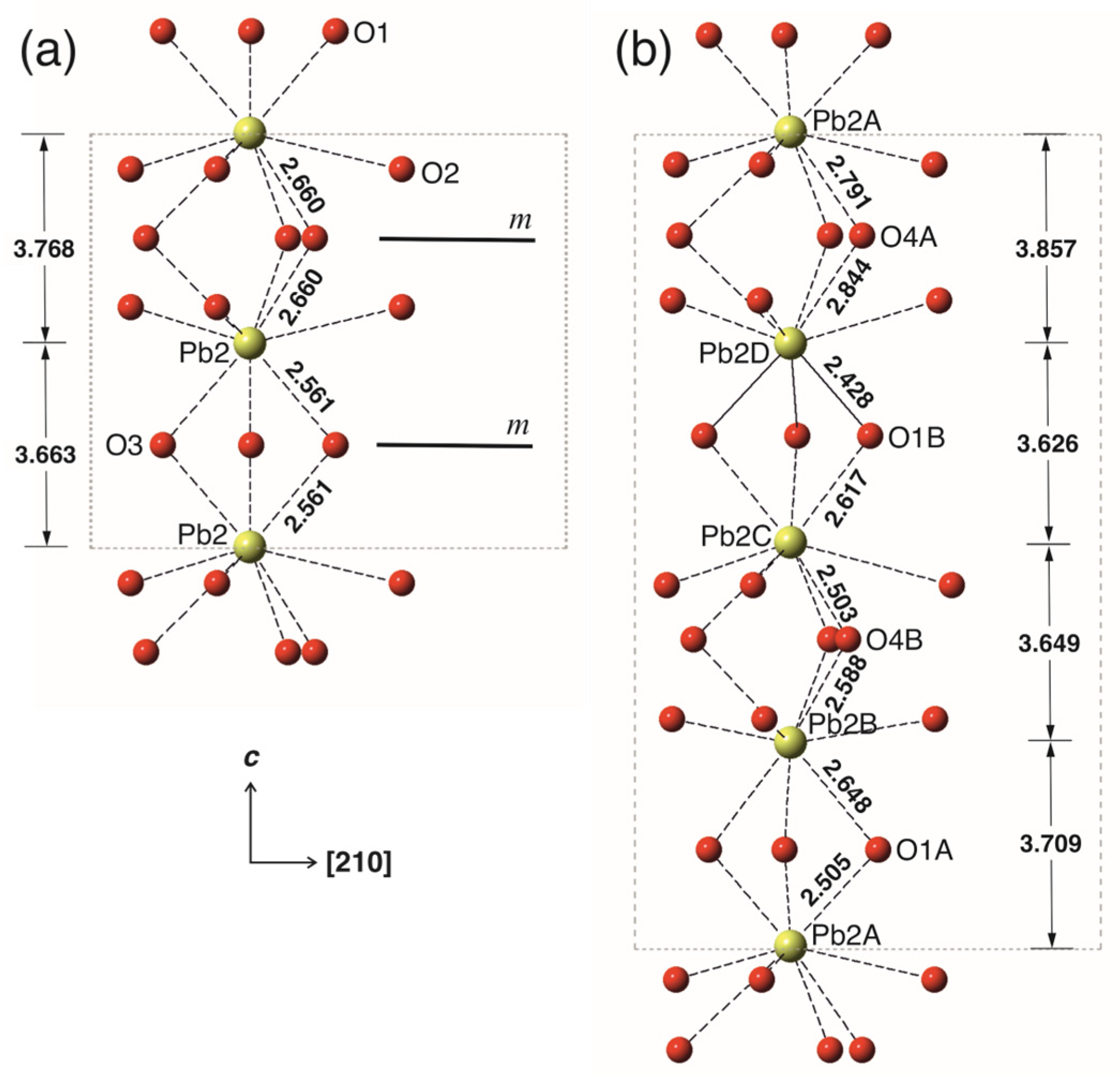{kind=link}
| Temperature/K | 293(2) |
| Crystal system | trigonal |
| Space group | P |
| a/Å | 9.8109(6) |
| c/Å | 14.8403(12) |
| Volume/Å3 | 1237.06(15) |
| Z | 2 |
| Dcalc, g/cm3 | 7.135 |
| μ/mm−1 | 68.270 |
| F(000) | 2220 |
| Crystal size/mm3 | 0.06 × 0.05 × 0.01 |
| Radiation | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 5.50 to 69.06 |
| Index ranges | −6 ≤ h ≤ 15, −15 ≤ k ≤ 11, −23 ≤ l ≤ 22 |
| Reflections collected | 11,777 |
| Independent reflections | 3456 [Rint = 0.0885, Rsigma = 0.0955] |
| Data/restraints/parameters | 3456/0/105 |
| Goodness-of-fit on F2 | 0.866 |
| Final R indices [I ≥ 2σ(I)] | R1 = 0.0413, wR2 = 0.0749 |
| Final R indices [all data] | R1 = 0.0544, wR2 = 0.0771 |
| Largest diff. peak/hole/e Å−3 | 4.010/−4.537 |
| Site | BVS * | BVS ** | x/a | y/b | z/c | Uiso |
|---|---|---|---|---|---|---|
| Pb1A | 1.90 | 1.98 | 0.25369(5) | −0.00015(5) | −0.12920(4) | 0.01239(10) |
| Pb1B | 1.93 | 1.97 | −0.00445(6) | 0.22423(5) | 0.37576(6) | 0.02123(12) |
| Pb2A | 2.05 | 2.06 | 1/3 | 2/3 | 0.00402(8) | 0.0123(3) |
| Pb2B | 2.07 | 2.07 | 1/3 | 2/3 | 0.25395(7) | 0.0132(3) |
| Pb2C | 2.04 | 2.06 | 1/3 | 2/3 | 0.49982(8) | 0.0187(3) |
| Pb2D | 2.05 | 2.07 | 1/3 | 2/3 | −0.25587(7) | 0.0125(3) |
| P1A | 4.94 | 4.94 | 0.3740(3) | 0.4021(3) | 0.3698(3) | 0.0078(5) |
| P1B | 4.90 | 4.90 | 0.0230(3) | −0.3765(3) | −0.1215(3) | 0.0084(5) |
| O1A | 1.99 | 2.00 | −0.1578(9) | −0.4786(9) | −0.1223(9) | 0.0131(11) |
| O1B | 2.00 | 2.02 | 0.4953(10) | 0.3468(9) | 0.3692(9) | 0.0131(11) |
| O2A | 1.90 | 1.90 | 0.0807(14) | −0.2676(14) | −0.2034(6) | 0.0121(19) |
| O2B | 1.99 | 1.99 | 0.2777(14) | 0.3560(13) | 0.2826(6) | 0.0121(19) |
| O3A | 1.94 | 1.93 | 0.2533(14) | 0.3211(14) | 0.4471(6) | 0.021(2) |
| O3B | 1.85 | 1.85 | 0.0869(16) | −0.2716(17) | −0.0366(6) | 0.021(2) |
| O4A | 2.11 | 2.15 | 0.0949(9) | −0.4866(10) | −0.1233(9) | 0.0166(12) |
| O4B | 2.06 | 2.07 | 0.4563(10) | 0.5820(10) | 0.3809(9) | 0.0166(12) |
| O5A *** | 1.43 | 1.55 | 0 | 0 | 0.330(3) | 0.031(7) |
| O5B **** | 1.70 | 1.87 | 0 | 0 | 0.395(3) | 0.031(7) |
| Pb1A–O4A | 2.234(8) | Pb2A–O1A | 2.505(11) 3x |
| Pb1A–O2B | 2.451(10) | Pb2A–O4A | 2.791(11) 3x |
| Pb1A–O2A | 2.554(12) | Pb2A–O3B | 2.835(13) 3x |
| Pb1A–O3B | 2.638(10) | <Pb2A–O> | 2.710 |
| Pb1A–O3B | 2.703(13) | ||
| Pb1A–O1A | 2.801(8) | Pb2B–O4B | 2.588(11) 3x |
| <Pb1A–O> | 2.564 | Pb2B–O1A | 2.647(11) 3x |
| Pb2B–O2B | 2.847(11) 3x | ||
| Pb1B–O5B | 2.240(6) | <Pb2B–O> | 2.694 |
| Pb1B–O5A | 2.325(14) | ||
| Pb1B–O3A | 2.453(12) | Pb2C–O4B | 2.503(11) 3x |
| Pb1B–O4B | 2.731(8) | Pb2C–O1B | 2.616(11) 3x |
| Pb1B–O3A | 2.749(10) | Pb2C–O3A | 3.172(12) 3x |
| Pb1B–O2A | 2.757(10) | <Pb2C–O> | 2.764 |
| Pb1B–O2B | 2.769(12) | ||
| Pb1B–O1B | 3.057(8) | Pb2D–O1B | 2.428(11) 3x |
| Pb1B–O3A | 3.175(12) | Pb2D–O4A | 2.843(11) 3x |
| <Pb1B–O> | 2.695 | Pb2D–O2A | 2.961(11) 3x |
| <Pb2D–O> | 2.744 | ||
| P1A–O2B | 1.532(11) | P1B–O2A | 1.527(11) |
| P1A–O1B | 1.536(9) | P1B–O1A | 1.541(8) |
| P1A–O4B | 1.539(9) | P1B–O3B | 1.548(12) |
| P1A–O3A | 1.552(12) | P1B–O4A | 1.558(9) |
| <P1A–O> | 1.540 | <P1B–O> | 1.544 |
| Phase | a, Å | c, Å | V/Z, Å3 | O’ Coordination | O’–Pb, Å | Ref. |
|---|---|---|---|---|---|---|
| Pb10(PO4)6O-WE | 9.84 | 14.86 | 623.0 | - | - | [39] |
| Pb10(PO4)6O-WE | 9.811 | 14.840 | 618.5 | trigonal | 2.240/2.325 3x | this work |
| Pb10(PO4)6O-KB | 9.865 | 7.431 | 626.3 | trigonal | 2.582 3x | [34] |
| Pb10(PO4)6(OH)2 | 9.866 | 7.426 | 626.0 | trigonal | 2.896 3x | [35] |
| Pb10(PO4)6(OH)2 | 9.883 | 7.441 | 629.4 | trigonal | 2.588 3x | [36] |
| Pb10(PO4)6(OH)2 | 9.774 | 7.291 | 603.2 | octahedral | 2.926 6x | [47] |
| Pb10(PO4)6(OH)2 | 9.871 | 7.427 | 626.7 | - | - | [48] |
| Pb10(PO4)6(OH)2 | 9.787 | 7.307 | 606.2 | trigonal | 2.708 3x | [49] |
| LK-99 | 9.843 | 7.428 | 623.2 | - | - | [3] |
| Pb9Cu(PO4)6O | 9.739 | 7.395 | 607.3 | - | - | [28] |
| Pb10−xCux(PO4)6O | 9.854 | 7.421 | 624.0 | - | - | [19] |
| Pb9Cu(PO4)6(OH)2 | 9.851 | 7.440 | 625.2 | - | - | [24] |
| Pb8Cu2(PO4)6(OH)2 | 9.870 | 7.398 | 624.1 | - | - | [48] |
| Pb6Cu4(PO4)6(OH)2 | 9.868 | 7.392 | 623.4 | - | - | [48] |
| Pb4Cu6(PO4)6(OH)2 | 9.866 | 7.383 | 622.3 | - | - | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krivovichev, S.V.; Engel, G. The Crystal Structure of Pb10(PO4)6O Revisited: The Evidence of Superstructure. Crystals 2023, 13, 1371. https://doi.org/10.3390/cryst13091371
Krivovichev SV, Engel G. The Crystal Structure of Pb10(PO4)6O Revisited: The Evidence of Superstructure. Crystals. 2023; 13(9):1371. https://doi.org/10.3390/cryst13091371
Chicago/Turabian StyleKrivovichev, Sergey V., and Günther Engel. 2023. "The Crystal Structure of Pb10(PO4)6O Revisited: The Evidence of Superstructure" Crystals 13, no. 9: 1371. https://doi.org/10.3390/cryst13091371
APA StyleKrivovichev, S. V., & Engel, G. (2023). The Crystal Structure of Pb10(PO4)6O Revisited: The Evidence of Superstructure. Crystals, 13(9), 1371. https://doi.org/10.3390/cryst13091371