
Red and Blue-Black Tin Monoxide, SnO: Pitfalls, Challenges, and Helpful Tools in Crystal Structure Determination of Low-Intensity Datasets from Microcrystals


Abstract
:1. Introduction
2. Materials and Methods
2.1. Blue-Black SnO
2.2. Red SnO
3. Results
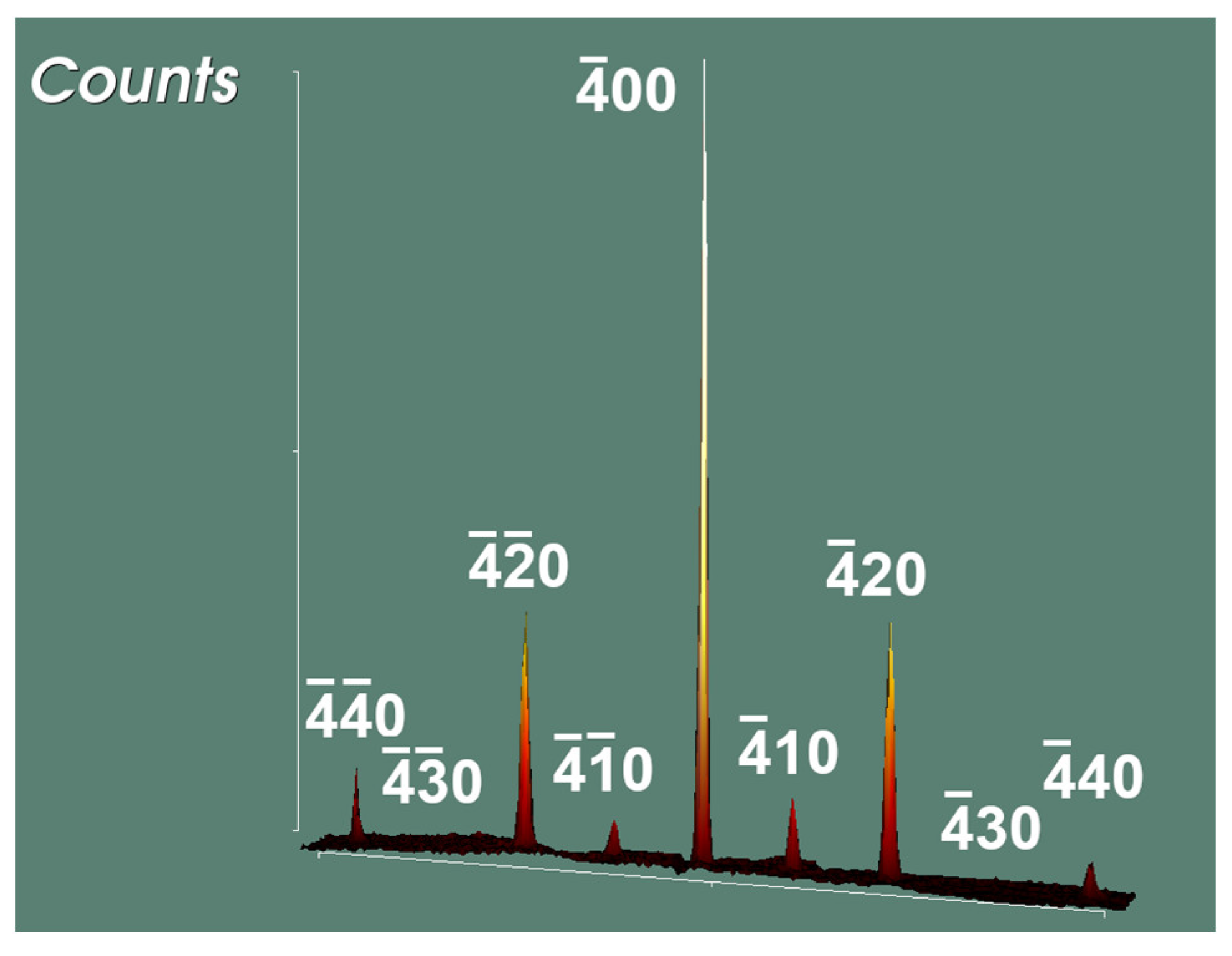
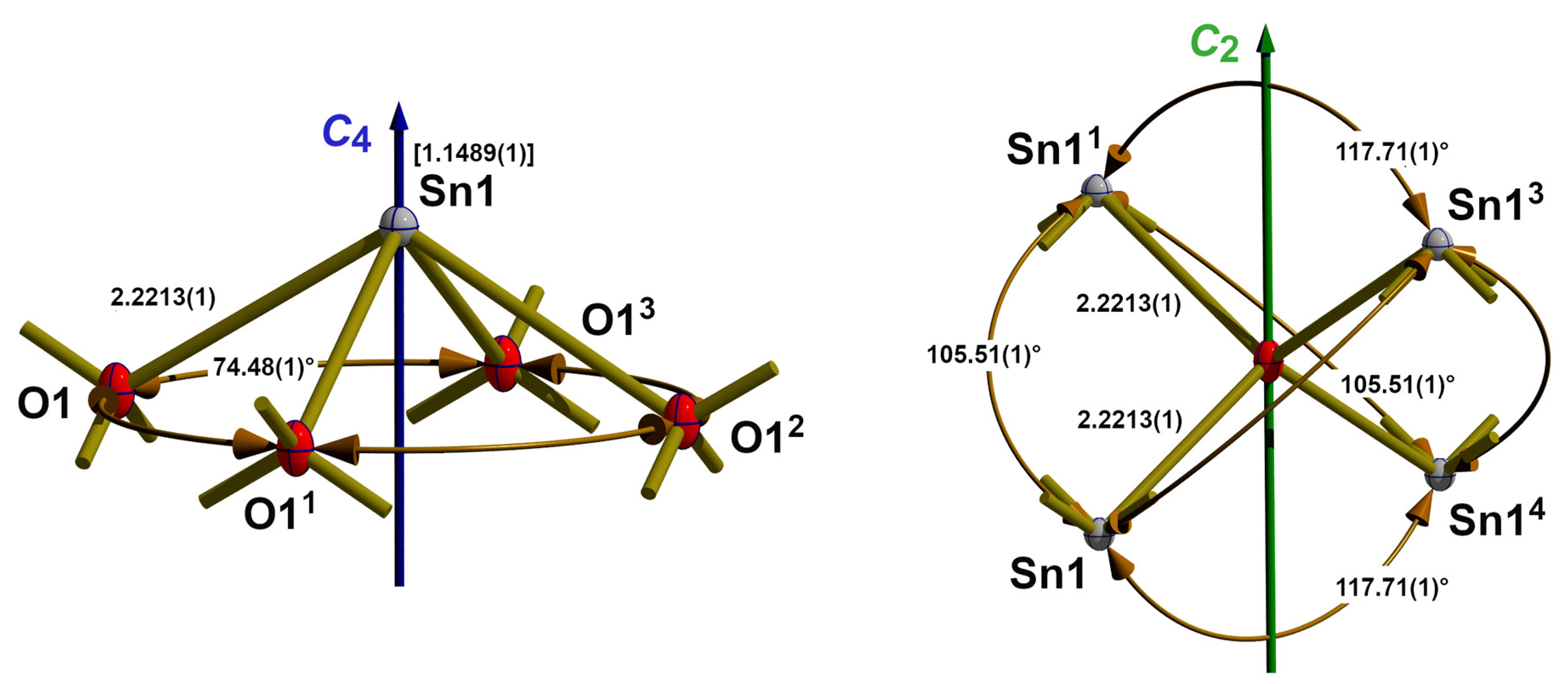
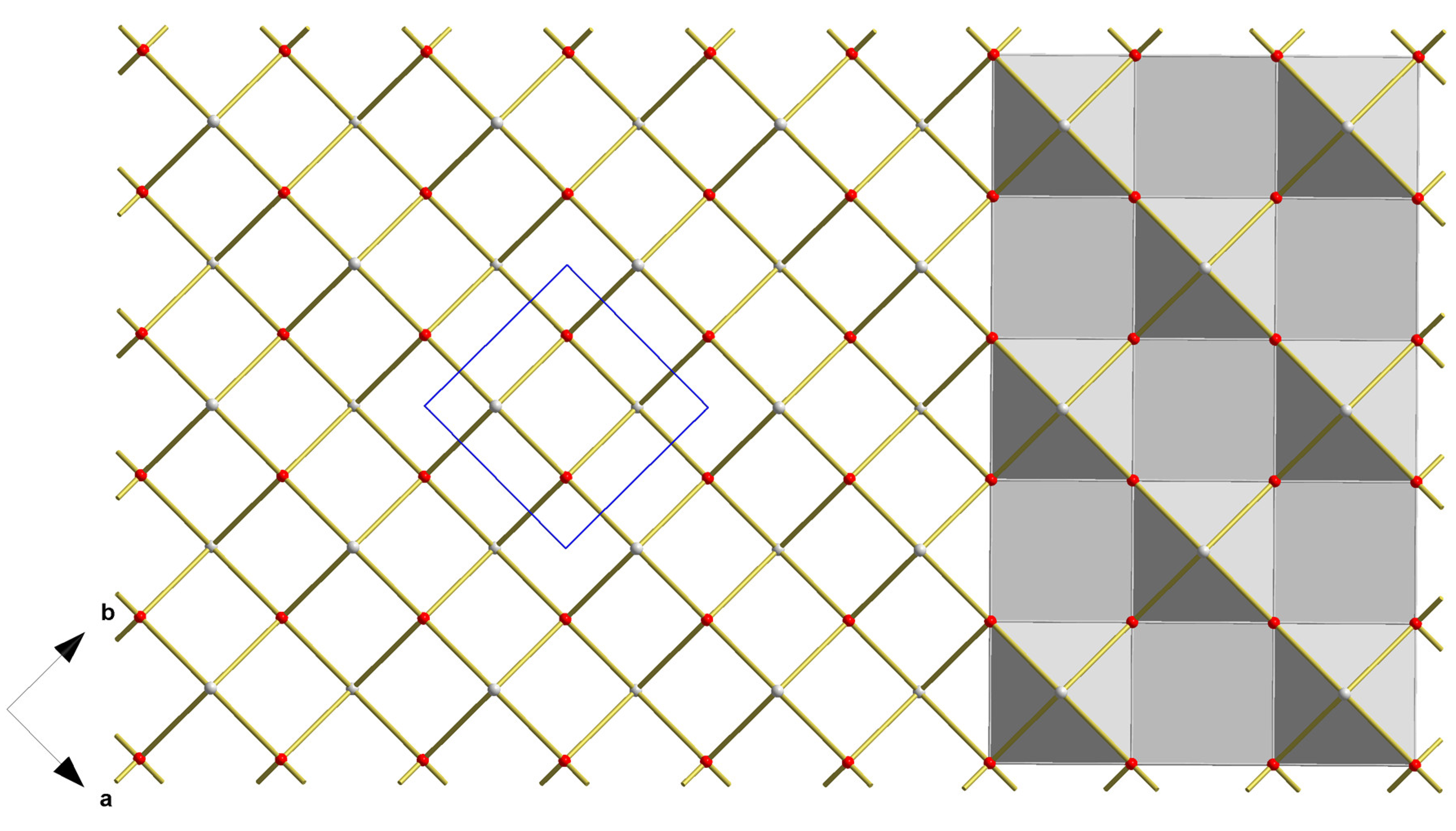
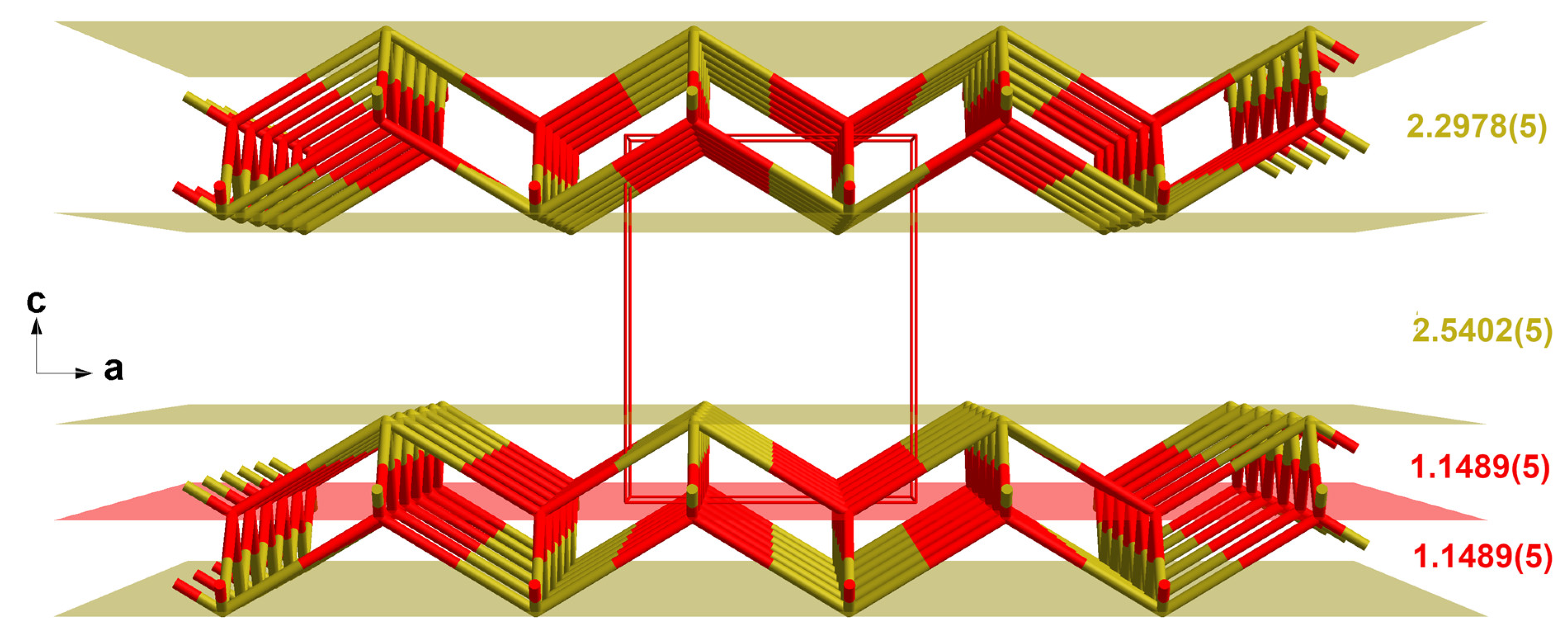
3.1. Blue-Black SnO
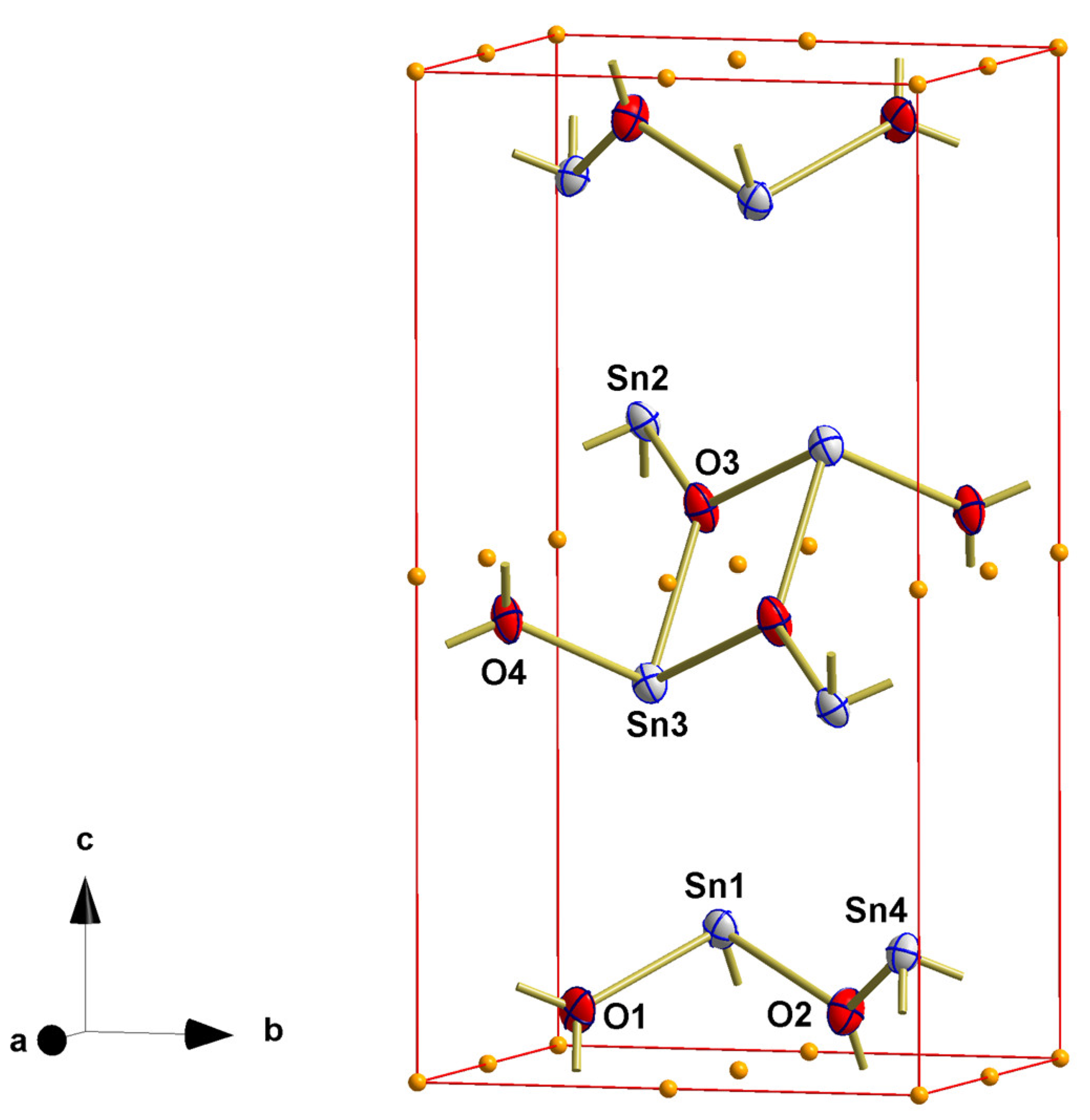
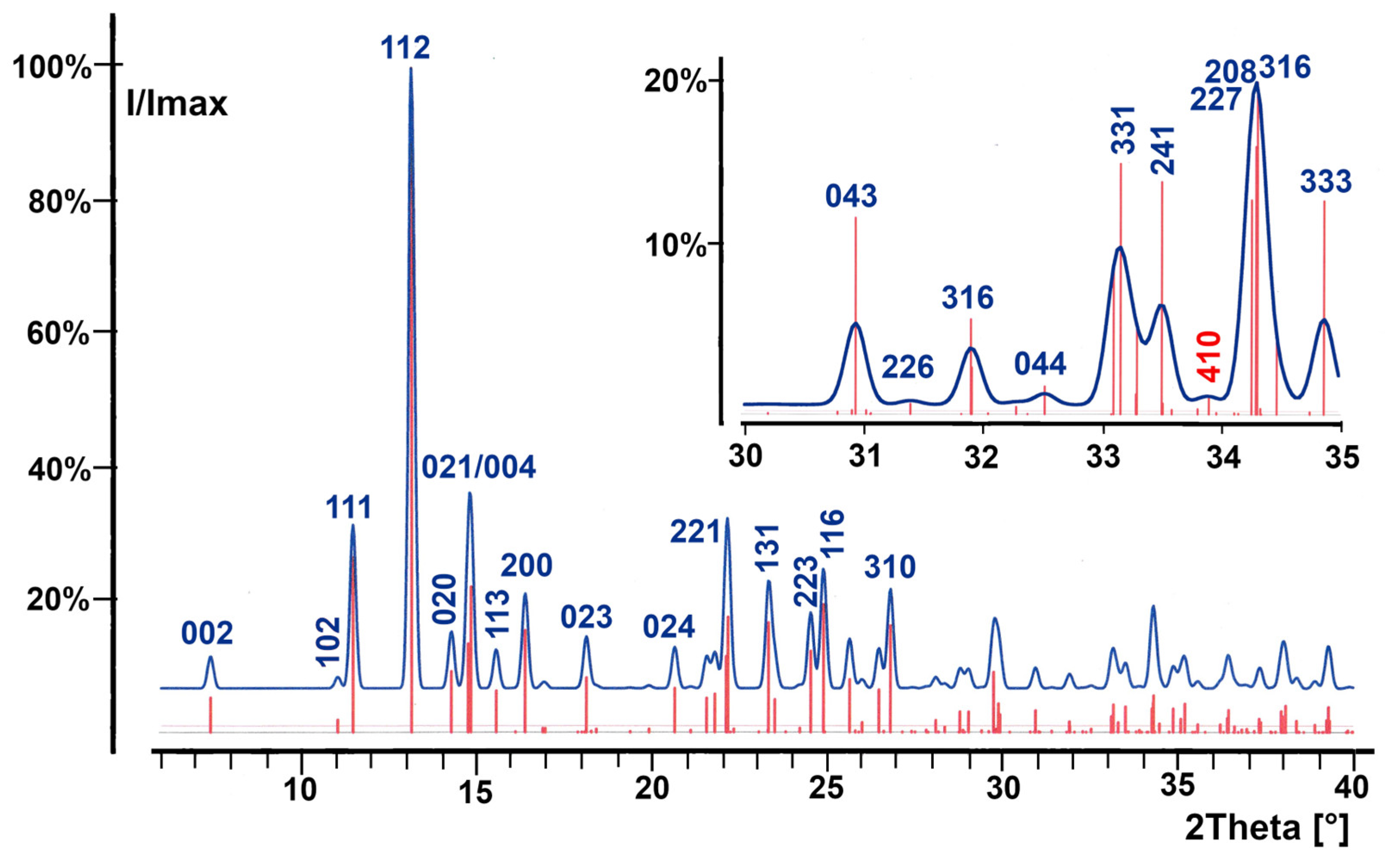
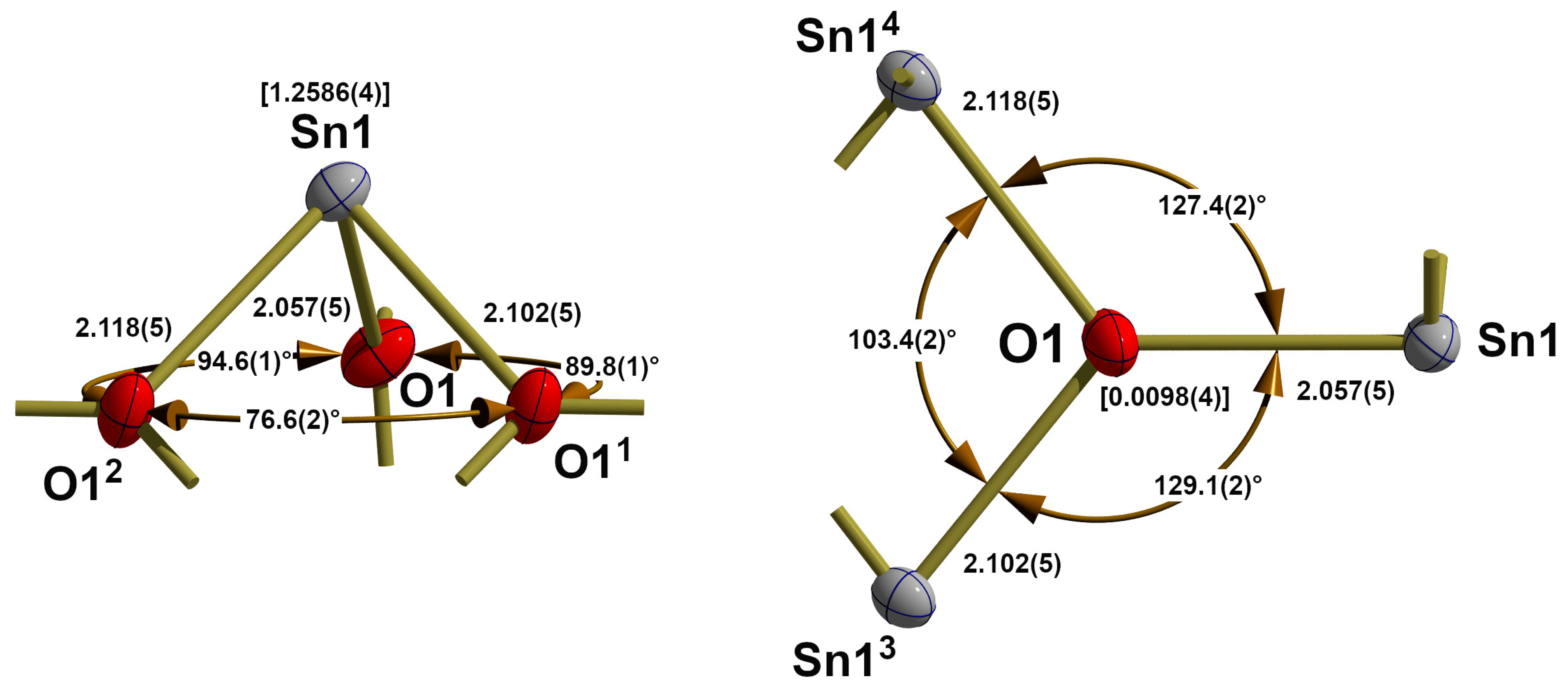
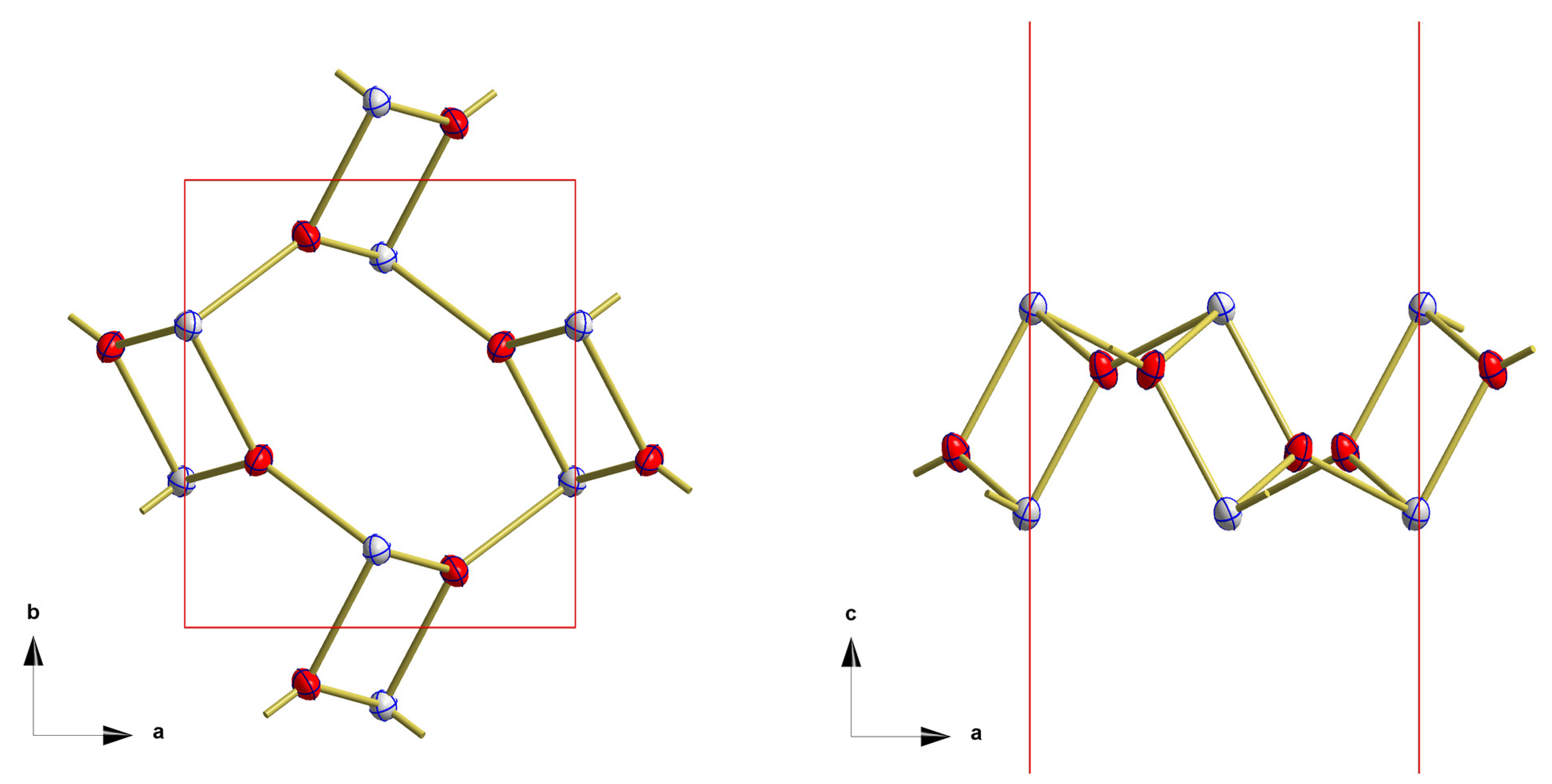
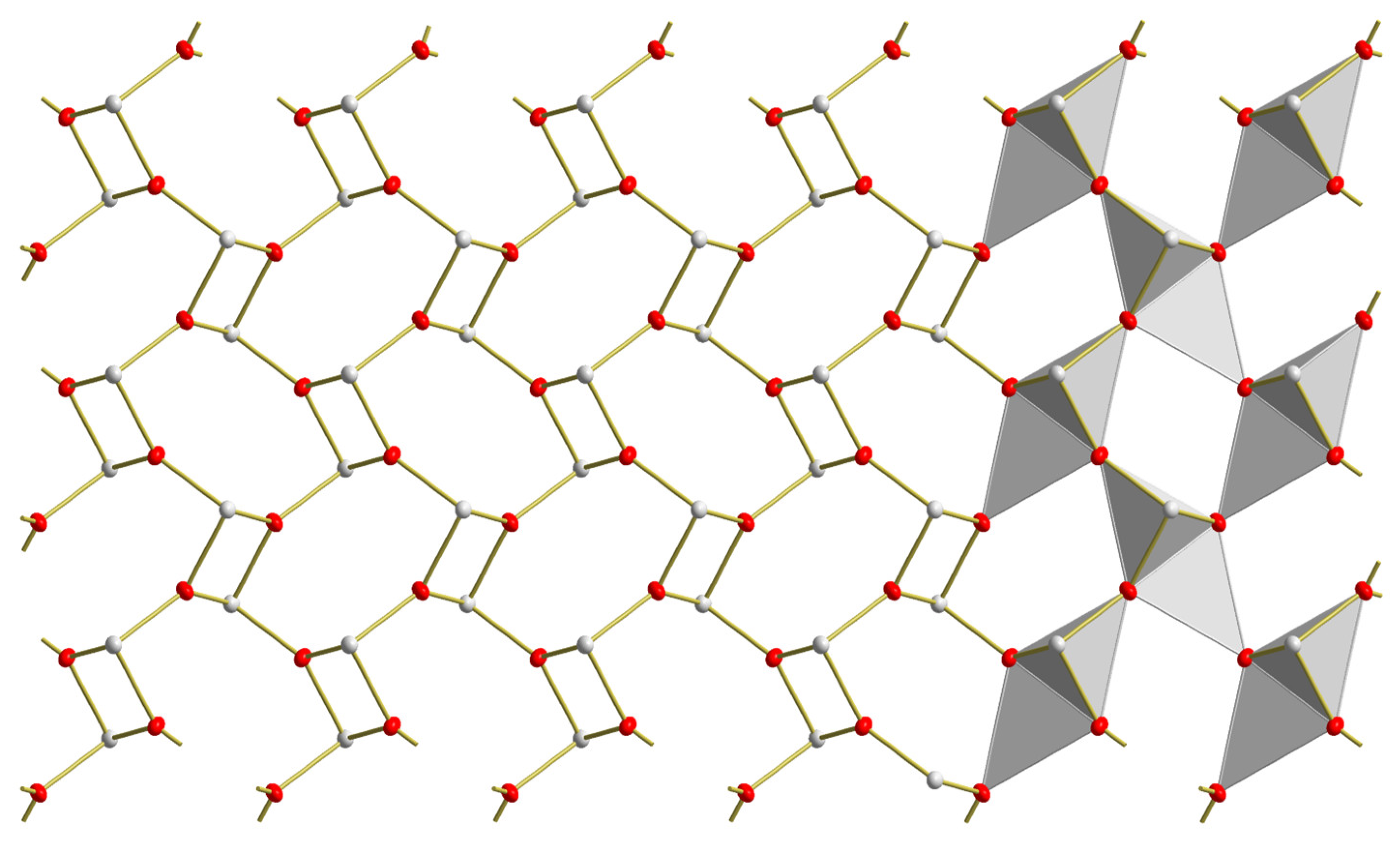
3.2. Red SnO
4. Experimental
5. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
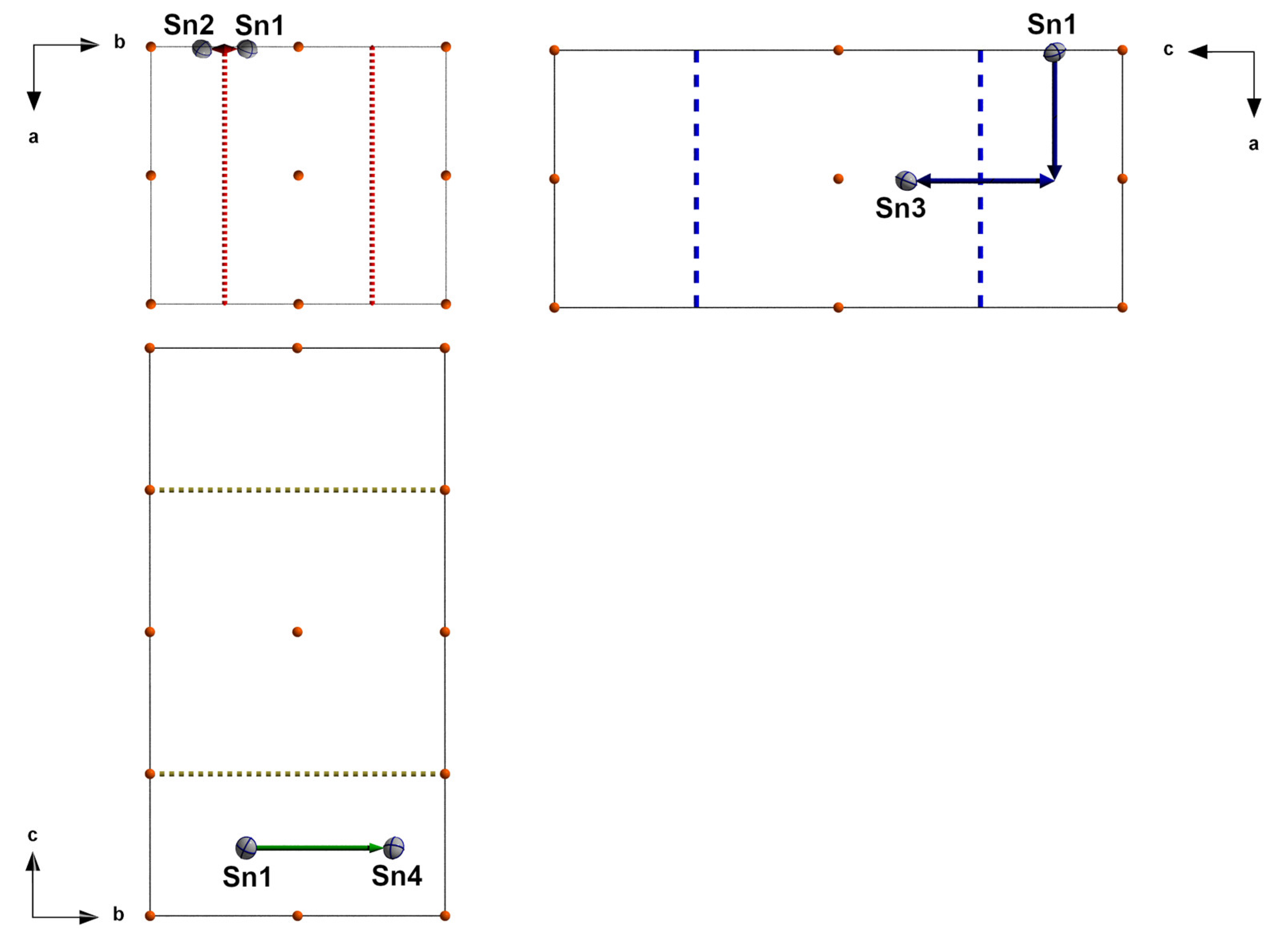
Appendix A. Manual Evaluation of Space Group Symmetry from the Triclinic Solution

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | Coordinates | Glide Planes | ||||
|---|---|---|---|---|---|---|
| x | y | z | operation | location | translation | |
| Sn(1) | 0.0084 = x | 0.3262 = y | 0.1194 = z | |||
| Sn(2) | 0.0084 | 0.1737 | 0.6194 | x, ½ − y, ½ + z | x, ¼, z | c |
| Sn(3) | 0.5083 | 0.3261 | 0.3807 | ½ + x, y, ½ − z | ¼, y, z | a |
| Sn(4) | 0.4915 | 0.8262 | 0.1194 | ½ − x, ½ + y, z | x, y, ¼ | b |
| O(1) | 0.3108 = x | 0.1257 = y | 0.0479 = z | |||
| O(3) | 0.3105 | 0.3741 | 0.5476 | x, ½ − y, ½ + z | x, ¼, z | c |
| O(2) | 0.1898 | 0.6264 | 0.0482 | ½ − x, ½ + y, z | x, y, ¼ | b |
| O(4) | 0.8124 | 0.1268 | 0.4528 | ½ + x, y, ½ − z | ¼, y, z | a |

References
- Proust, J.L. Recherche sur l’étain. J. Phys. Chim. Hist. Nat. Arts 1800, 51, 173–184. [Google Scholar]
- Berzelius, J.J. Des oxides d’étain et de tellure. Ann. Chim. Ser. 1 1813, 87, 50–97. [Google Scholar]
- Gay-Lussac, J.L. Sur l’Oxidation de quelques métaux. Ann. Chim. Phys. Ser. 2 1816, 1, 32–45. [Google Scholar]
- Frémy, E. Recherches sur les acides métalliques. Compt. Rend. Hebd. 1842, 15, 1106–1111. [Google Scholar]
- Roth, R. Ueber das rothe Zinnoxydul. Jahrb. Prakt. Pharm. 1845, 10, 381–382. [Google Scholar]
- Weiser, H.B.; Milligan, W.O. X_Ray Studies on the Hydrous Oxides III. Stannous Oxid. J. Phys. Chem. 1932, 32, 3039–3045. [Google Scholar] [CrossRef]
- Levi, G.R. Isomorfismo degli ossidi stannoso e piomboso. Nuovo Cimento 1924, 1, 335–346. [Google Scholar] [CrossRef]
- Straumanis, M.; Strenk, C. Über das Zinn(2)-Oxyd. Z. Anorg. Allg. Chem. 1933, 213, 301–309. [Google Scholar] [CrossRef]
- Moore, W.J.; Pauling, L. The Crystal Structure of the Tetragonal Monoxides of Lead, Tin, Palladium, and Platinum. J. Am. Chem. Soc. 1941, 63, 1392–1394. [Google Scholar] [CrossRef]
- Pannetier, J.; Denes, G. Tin(II) Oxide: Structure Refinement and Thermal Expansion. Acta Cyrstallogr. 1980, B36, 2763–2765. [Google Scholar] [CrossRef]
- Izumi, F. Pattern-Fitting Structure Refinement of Tin(II) Oxide. J. Sol. State Chem. 1981, 38, 381–385. [Google Scholar] [CrossRef]
- Herber, R.H. Mössbauer lattice temperature of tetragonal (P4/nmm) SnO. Phys. Rev. 1983, B27, 4013–4017. [Google Scholar] [CrossRef]
- Moreno, M.S.; Mercader, R.C. Mössbauer study of SnO lattice dynamics. Phys. Rev. 1994, B50, 9875–9881. [Google Scholar] [CrossRef] [PubMed]
- Boher, P.; Garnier, P.; Gavarri, J.R.; Hewat, A.W. Monoxyde quadratique PbOα(I): Description de la transition structurale ferroe´lastique. J. Solid State Chem. 1985, 57, 343–350. [Google Scholar] [CrossRef]
- Organ, R.M.; Mandarino, J.A. Romarchite and hydroromarchite, two new stannous minerals (Abstract). Can. Mineral. 1971, 10, 916. [Google Scholar]
- Warr, L.N. IMA-CNMNC approved mineral symbols. Miner. Mag. 2021, 85, 291–320. [Google Scholar] [CrossRef]
- Donaldson, J.D.; Moser, W.; Simpson, W.B. Red Tin(II) Oxide. J. Chem. Soc. 1961, 839–841. [Google Scholar] [CrossRef]
- Hill, P.A. Refinement of the structure of orthorhombic PbO (massicot) by Rietveld analysis of neutron powder diffraction data. Acta Cryst. 1985, C41, 1281–1284. [Google Scholar] [CrossRef]
- Donaldson, J.D.; Moser, W.; Simpson, W.B. The structure of the red modification of tin(II) oxide. Acta Cryst. 1963, 16, A22. [Google Scholar]
- Köhler, J.; Tong, J.; Dinnebier, R.E.; Simon, A. Crystal Structure and Electronic Structure of Red SnO. Z. Anorg. Allg. Chem. 2012, 638, 1970–1975. [Google Scholar] [CrossRef]
- Wittmann, S.; Murshed, M.M.; Gesing, T.M. On red tin (II) oxide: Temperature-dependent structural, spectroscopic, and thermogravimetric properties. Z. Anorg. Allg. Chem. 2022, 648, e202200311. [Google Scholar] [CrossRef]
- Gagné, O.C.; Hawthorne, F.C. Comprehensive derivation of bond-valence parameters for ion pairs involving oxygen. Acta Cryst. 2015, B71, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.; Watson, G.W. Electronic structures of rocksalt, litharge, and herzbergite SnO by density functional theory. Phys. Rev. 2004, B70, 235114. [Google Scholar] [CrossRef]
- Allen, J.P.; Scanlon, D.O.; Parker, S.C.; Watson, G.W. Tin Monoxide: Structural Prediction from First Principles Calculations with van der Waals Corrections. J. Phys. Chem. C 2011, 115, 19916–19924. [Google Scholar] [CrossRef]
- Zhou, W.; Umezawa, N. Band gap engineering of bulk and nanosheet SnO: An insight into the interlayer Sn-Sn lone pair interactions. Phys. Chem. Chem. Phys. 2015, 17, 17816–17820. [Google Scholar] [CrossRef] [PubMed]
- Mubeen, A.; Majid, A.; Alkhedher, M.; Tag-ElDin, E.M.; Bulut, N. Structural and Electronic Properties of SnO Downscaled to Monolayer. Materials 2022, 15, 5578. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, J.-Q.; Blatov, V.A.; Gong, Y.; Umezawa, N.; Tada, T.; Hosono, H.; Oganov, A.R. Crystal and electronic structure engineering of tin monoxide by external pressure. J. Adv. Ceram. 2021, 10, 565–577. [Google Scholar] [CrossRef]
- Allen, J.P.; Scanlon, D.O.; Piper, L.F.J.; Watson, G.W. Understanding the defect chemistry of tin monoxide. J. Mater. Chem. C 2013, 1, 8184–8208. [Google Scholar] [CrossRef]
- Miller, S.A.; Gorai, P.; Aydemir, U.; Mason, T.O.; Stevanović, V.; Toberer, E.S.; Snyder, G.F. SnO as potential thermoelectric candidate. J. Mater. Chem. C 2017, 5, 8854–8861. [Google Scholar] [CrossRef]
- Hahn, T. International Tables for Crystallography, 4th ed.; Vol. A—Space-Group Symmetry; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Bruker. APEX 3—Suite of Crystallographic Software, 2015.9, ed.; Bruker: Madison, Wisconsin, USA, 2015. [Google Scholar]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Cryst. 2020, E76, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Puff, H.; Reuter, H. Zur Hydrolyse von Monoorganylzinn-trihalogeniden III. Isolierung und Röntgenstrukturanalyse von Verbindungen mit dem neuartigen Käfig-Ion [(i-PrSn)12O14(OH)6]2+. J. Organomet. Chem. 1989, 373, 173–184. [Google Scholar] [CrossRef]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef] [PubMed]
- Schnering, H.G.V.; Nesper, R.; Pelshenke, H. Hydroxoverbindungen. 10. Über die Natriumoxohydroxostannate(II) Na4[Sn4O(OH)10] und Na2[Sn2O(OH)4]. Z. Anorg. Allg. Chem. 1983, 499, 117–129. [Google Scholar] [CrossRef]
- Nesper, R.; von Schnering, H.G. Barium-Oxohydroxostannat(II) Ba[SnO(OH)]2. Z. Anorg. Allg. Chem. 1983, 499, 109–116. [Google Scholar] [CrossRef]
- Bolzan, A.A.; Fong, C.; Kennedy, B.J.; Howard, C.J. Structural Studies of Rutile-Type Metal Dioxides. Acta Cryst. 1977, B53, 373–380. [Google Scholar] [CrossRef]
- Bruker. SAINT, 7.56a; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Bruker. SADABS, 2008.1; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Wilson, A.J.C. (Ed.) International Tables for Crystallography; Kluwer Academic Publishers: Dordrecht, The Netherland, 1992; Volume C. [Google Scholar]
- Brandenburg, K. Diamond—Crystal and Molecular Visualization; Crystal Impact: Bonn, Germany, 2006. [Google Scholar]
- POV-Ray—Persistence of Vision Raytracer, Version 3.7; Persistence of Vision Pty. Ltd.: Williamstown, VIC, Australia, 2004; Available online: http://www.povray.org/download/ (accessed on 31 March 2014).










| Modification | Blue-Black | Red | |
|---|---|---|---|
| Empirical formula | SnO | ||
| Formula weight | 134.69 | ||
| Crystallographic data | |||
| Temperature (K) | 100(2) | 296(2) | 296(2) |
| Crystal system | tetragonal | tetragonal | orthorhombic |
| Space group | P4/nmm | P4/nmm | Pbca |
| a (Å) | 3.7922(3) | 3.8021(1) | 5.0116(3) |
| b (Å) | 3.7922(3) | 3.8021(1) | 5.7445(4) |
| c (Å) | 4.8025(5) | 4.8381(1) | 11.0600(8) |
| V (Å3) | 69.06(1) | 69.939(4 | 318.41(4) |
| Z, Z’ | 2, ⅛ | 2, ⅛ | 8, 1 |
| dcalc (g/cm3) | 6.477 | 6.396 | 5.619 |
| μ(MoKα) (mm−1) | 17.788 | 17.565 | 15.433 |
| F(000) | 116 | 116 | 464 |
| Data collection | |||
| 2Θmax | 58° | 72° | 58° |
| Time/frame (s) | 10 | 40 | 40 |
| Total exposure time (h) | 15.54 | 125.64 | 106.75 |
| Total number of frames | 6540 | 11,308 | 9453 |
| Reflections collected/unique | 3757/77 | 11,651/120 | 26,197/422 |
| Rint | 0.0287 | 0.0168 | 0.0758 |
| Rσ | 0.0064 | 0.0035 | 0.0124 |
| Refinement | |||
| Data/restraints/parameters | 77/0/7 | 120/0/6 | 422/0/19 |
| Goof | 1.423 | 1.306 | 1.087 |
| R1, wR2 (I > 2σ(I)) | 0.0079/0.0224 | 0.0081/0.0176 | 0.0274/0.0644 |
| R1, wR2 (all data) | 0.0080/0.0225 | 0.0086/0.0177 | 0.0398/0.0725 |
| Weighting factors a/b | 0.0084/0.1036 | 0.0066/0.0576 | 0.0318/3.9846 |
| Extinction coefficient | 0.030(5) | n/a | n/a |
| ±Δe (e/Å3) | 0.444/−0.480 | 0.575/−0.445 | 1.928/−0.973 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reuter, H. Red and Blue-Black Tin Monoxide, SnO: Pitfalls, Challenges, and Helpful Tools in Crystal Structure Determination of Low-Intensity Datasets from Microcrystals. Crystals 2023, 13, 1281. https://doi.org/10.3390/cryst13081281
Reuter H. Red and Blue-Black Tin Monoxide, SnO: Pitfalls, Challenges, and Helpful Tools in Crystal Structure Determination of Low-Intensity Datasets from Microcrystals. Crystals. 2023; 13(8):1281. https://doi.org/10.3390/cryst13081281
Chicago/Turabian StyleReuter, Hans. 2023. "Red and Blue-Black Tin Monoxide, SnO: Pitfalls, Challenges, and Helpful Tools in Crystal Structure Determination of Low-Intensity Datasets from Microcrystals" Crystals 13, no. 8: 1281. https://doi.org/10.3390/cryst13081281
APA StyleReuter, H. (2023). Red and Blue-Black Tin Monoxide, SnO: Pitfalls, Challenges, and Helpful Tools in Crystal Structure Determination of Low-Intensity Datasets from Microcrystals. Crystals, 13(8), 1281. https://doi.org/10.3390/cryst13081281