
Lead-Free Double Perovskites: A Review of the Structural, Optoelectronic, Mechanical, and Thermoelectric Properties Derived from First-Principles Calculations, and Materials Design Applicable for Pedagogical Purposes

 , , , , , and
, , , , , and
Abstract
:1. Introduction
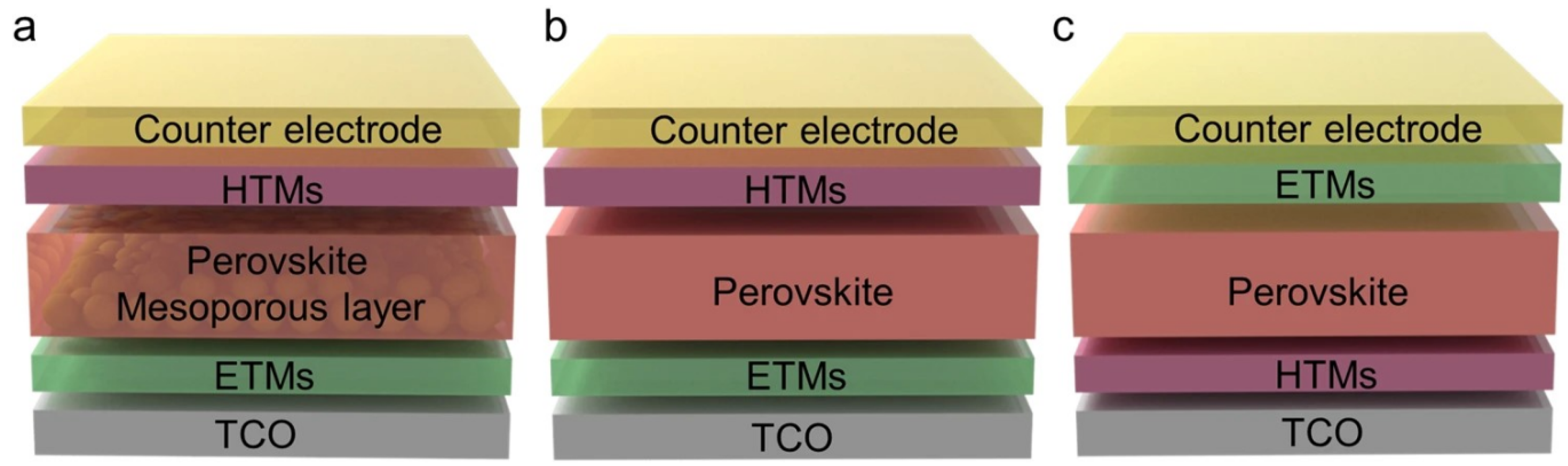
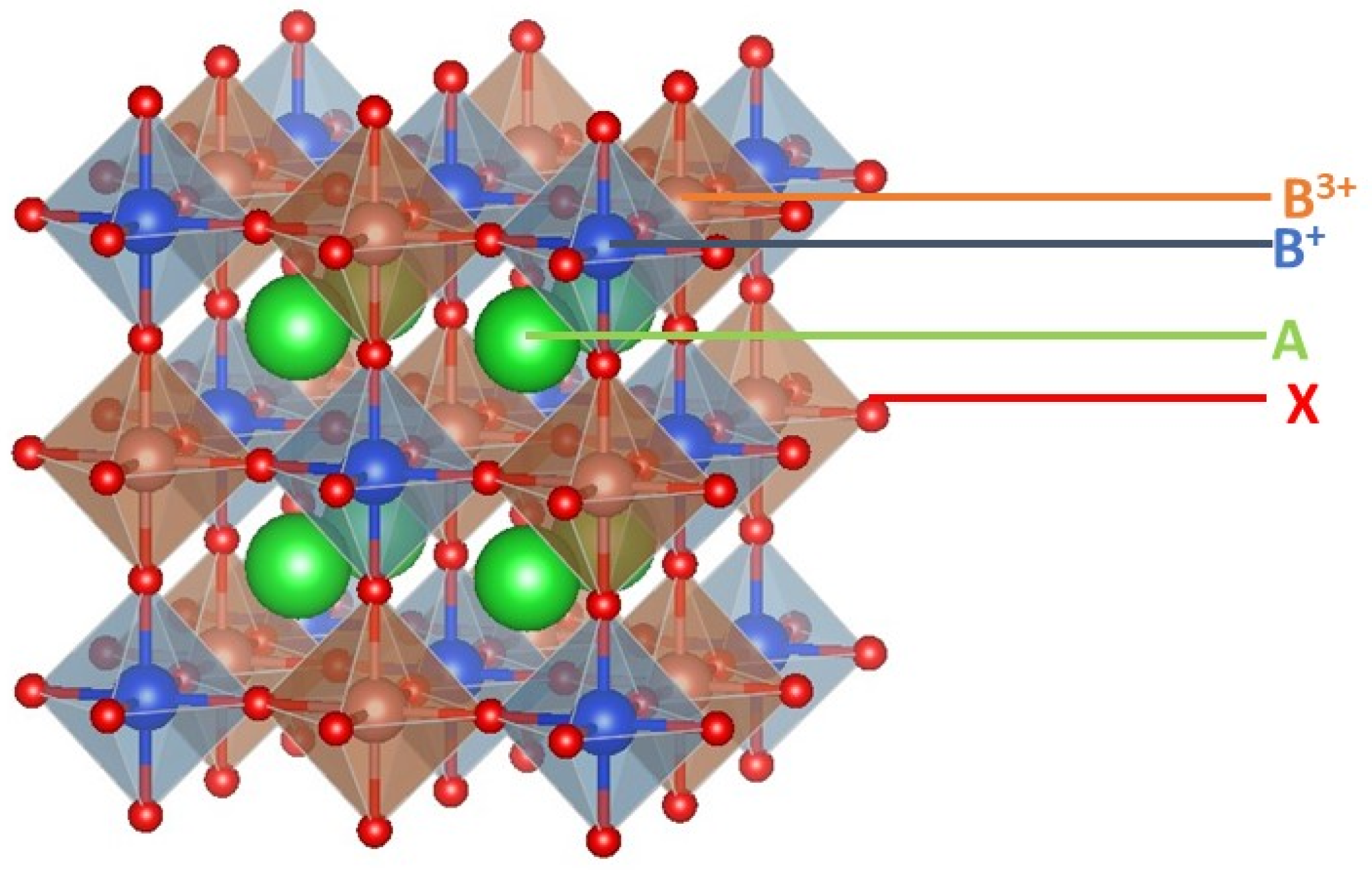
2. Structural and Electronics Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | Materials | Lattice Parameter Å | Bandgap (eV) | Bandgap Type | Functionals | Ref. |
|---|---|---|---|---|---|---|
| 1 | Na2ScAgCl6 | 10.19 | 3.63 | direct | PBE | [68] |
| 2 | Na2CuSbCl6 | 10.29 | 0.66 | indirect | PBE | [72] |
| 3 | Na2CuBiCl6 | 10.41 | 0.94 | indirect | PBE | [72] |
| 4 | K2ScAgCl6 | 10.26 | 3.65 | direct | PBE | [68] |
| 5 | K2CuBiCl6 | 10.47 | 1.44 | indirect | HSE06 | [73] |
| 6 | K2CuBiBr6 | 11.05 | 1.03 | indirect | HSE06 | [73] |
| 7 | K2AgInCl6 | 10.32 | 2.37 | direct | TB-Mbj | [69] |
| 8 | K2AgInBr6 | 10.86 | 1.34 | direct | TB-Mbj | [69] |
| 9 | K2AgInI6 | 11.79 | 0.00 | direct | TB-Mbj | [69] |
| 10 | K2AgSbCl6 | 10.66 | 2.30 | indirect | mBJ | [50] |
| 11 | K2AgSbBr6 | 11.22 | 1.51 | indirect | mBJ | [50] |
| 12 | K2AgSbI6 | 12.01 | 0.70 | indirect | mBJ | [50] |
| 13 | K2AgBiI6 | 12.11 | 1.21 | indirect | mBJ | [75] |
| 14 | K2InAgCl6 | 10.66 | 1.12 | direct | PBE | [70] |
| 15 | Rb2KScI6 | 12.15 | 2.75 | direct | mBJ | [75] |
| 16 | Rb2InAgCl6 | 10.66 | 1.11 | direct | PBE | [70] |
| 17 | Rb2TlAgCl6 | 10.45 | 0.78 | direct | TB-Mbj | [76] |
| 18 | Cs2NaSbCl6 | 10.65 | 3.13 | indirect | PBE | [74] |
| 19 | Cs2NaSbBr6 | 11.22 | 2.54 | indirect | PBE | [74] |
| 20 | Cs2NaSbI6 | 12.06 | 1.90 | direct | PBE | [74] |
| 21 | Cs2NaBiCl6 | 10.84 | 3.73 | indirect | PBE | [74] |
| 22 | Cs2NaBiBr6 | 11.36 | 3.07 | indirect | PBE | [74] |
| 23 | Cs2NaBiI6 | 12.20 | 2.23 | direct | PBE | [74] |
| 24 | Cs2KScI6 | 12.17 | 2.65 | direct | mBJ | [75] |
| 25 | Cs2AgCrCl6 | 10.17 | 0.00 | HSE06 | [33] | |
| 26 | Cs2AgFeCl6 | 10.48 | 1.18 | indirect | mBJ | [67] |
| 27 | Cs2AgCoCl6 | 10.21 | 2.34 | indirect | HSE06 | [33] |
| 28 | Cs2AgCoBr6 | 10.73 | 1.66 | indirect | HSE06 | [33] |
| 29 | Cs2AgCoI6 | 11.51 | 0.78 | indirect | HSE06 | [33] |
| 30 | Cs2AgRhCl6 | 10.32 | 2.42 | direct | HSE06 | [33] |
| 31 | Cs2AgRhBr6 | 10.83 | 1.85 | indirect | HSE06 | [33] |
| 32 | Cs2AgRhI6 | 11.59 | 0.85 | indirect | HSE06 | [33] |
| 33 | Cs2AgIrCl6 | 10.35 | 2.40 | direct | HSE06 | [33] |
| 34 | Cs2AgIrBr6 | 10.88 | 1.96 | direct | HSE06 | [33] |
| 35 | Cs2AgIrI6 | 11.61 | 1.27 | indirect | HSE06 | [33] |
| 36 | Cs2InCoCl6 | 10.47 | 0.00 | HSE06 | [33] | |
| 37 | Cs2InAgCl6 | 10.69 | 1.07 | direct | PBE | [70] |
| 38 | Cs2InBiCl6 | 11.27 | 1.84 | indirect | TB-Mbj | [71] |
| 39 | Cs2InBiBr6 | 11.71 | 1.27 | indirect | TB-Mbj | [71] |
| 40 | Cs2InBiI6 | 12.41 | 0.65 | indirect | TB-Mbj | [71] |
| 41 | Cs2TlAgCl6 | 10.52 | 0.74 | direct | TB-Mbj | [68] |
3. Optical Properties
4. Thermoelectric Properties
5. Mechanical Properties
| S/N | Materials | Nature of the Material | Pugh Ratio | Poisson Ratio | Ref. |
|---|---|---|---|---|---|
| 1 | Na2CuSbCl6 | ductile | 2.90 | 0.35 | [72] |
| 2 | Na2CuBiCl6 | ductile | 2.42 | 0.32 | [72] |
| 3 | K2CuBiCl6 | ductile | 4.04 | 0.39 | [73] |
| 4 | K2CuBiBr6 | ductile | 2.80 | 0.34 | [73] |
| 5 | K2AgSbCl6 | ductile | 2.45 | 0.32 | [50] |
| 6 | K2AgSbBr6 | ductile | 6.69 | 0.42 | [50] |
| 7 | Rb2KScI6 | brittle | 1.27 | 0.19 | [66] |
| 8 | Cs2KScCI6 | ductile | 2.07 | 0.29 | [87] |
| 9 | Cs2KScBr6 | ductile | 2.30 | 0.31 | [87] |
| 10 | Cs2KScI6 | brittle | 1.57 | 0.25 | [66] |
| 11 | Cs2AgFeCl6 | ductile | 2.17 | 0.30 | [67] |
| 12 | Cs2AgCoCl6 | ductile | 3.41 | 0.37 | [33] |
| 13 | Cs2AgCoBr6 | ductile | 2.61 | 0.33 | [33] |
| 14 | Cs2AgCoI6 | ductile | 2.81 | 0.34 | [33] |
| 15 | Cs2AgRhCl6 | ductile | 2.80 | 0.34 | [33] |
| 16 | Cs2AgRhBr6 | ductile | 2.60 | 0.33 | [33] |
| 17 | Cs2AgRhI6 | ductile | 2.54 | 0.33 | [33] |
| 18 | Cs2AgIrCl6 | ductile | 2.84 | 0.34 | [33] |
| 19 | Cs2AgIrBr6 | ductile | 2.73 | 0.34 | [33] |
| 20 | Cs2AgIrI6 | ductile | 3.05 | 0.35 | [33] |
6. Design and Discovery of Double Perovskite Materials Using Machine Learning (ML) and Databases to Teach the Concepts
6.1. Machine Learning for Materials Discovery

6.2. Databases to Teach the Concepts of Materials Discovery
7. Conclusions
- Studies suggest that material bandgaps can be altered through metal doping, alloying, or introducing defects. Further research is crucial to understand the relationship between crystal structure, lattice constants, and bandgaps in Pb-free halide double perovskites, which will aid in developing tailored materials for specific electronic and optical applications.
- Double perovskites display favourable optical absorption for photovoltaic applications. Certain compounds with indirect bandgaps could find use in UV sensors. Additionally, the strong ductility of most of the compounds investigated in this review suits diverse industrial applications such as in catalysis, fuel cells, and electrochemical sensing.
- Although Pb-free halide double perovskites are extensively studied for thermoelectric purposes, certain groups remain inadequately explored. This review proposes utilizing a robust computational screening approach by employing DFT and machine learning methods to fill these gaps in understanding.
- ML holds great potential in materials discovery and understanding the relationship between structural, compositional, technical characteristics, and performance. However, its use in material study is still in its early stages and requires further research to improve our understanding.
- The systematic data extraction techniques discussed in this review allow users to access valuable resources, enhance experiential learning, and develop proficiency in materials science.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kim, G.-H.; Kim, D.S. Development of Perovskite Solar Cells with >25% Conversion Efficiency. Joule 2021, 5, 1033–1035. [Google Scholar] [CrossRef]
- Dixit, S. Solar Technologies and Their Implementations: A Review. Mater. Today Proc. 2020, 28, 2137–2148. [Google Scholar] [CrossRef]
- Glunz, S.; Preu, R.; Biro, D. Crystalline Silicon Solar Cells: State-of-the-Art and Future Developments. In Comprehensive Renewable Energy; Elsevier: Amsterdam, Netherlands, 2012; Volume 1, pp. 353–387. ISBN 978-0-08-087873-7. [Google Scholar]
- Kung, P.-K.; Li, M.-H.; Lin, P.-Y.; Jhang, J.-Y.; Pantaler, M.; Lupascu, D.C.; Grancini, G.; Chen, P. Lead-Free Double Perovskites for Perovskite Solar Cells. Solar RRL 2020, 4, 1900306. [Google Scholar] [CrossRef]
- Jebakumar, J.P.A.; Moni, D.J.; Gracia, D.; Shallet, M.D. Design and Simulation of Inorganic Perovskite Solar Cell. Appl. Nanosci. 2022, 12, 1507–1518. [Google Scholar] [CrossRef]
- Meyer, E.; Mutukwa, D.; Zingwe, N.; Taziwa, R. Lead-Free Halide Double Perovskites: A Review of the Structural, Optical, and Stability Properties as Well as Their Viability to Replace Lead Halide Perovskites. Metals 2018, 8, 667. [Google Scholar] [CrossRef]
- Li, X.; Zhao, H.; Liang, J.; Luo, Y.; Chen, G.; Shi, X.; Lu, S.; Gao, S.; Hu, J.; Liu, Q. A-Site Perovskite Oxides: An Emerging Functional Material for Electrocatalysis and Photocatalysis. J. Mater. Chem. A 2021, 9, 6650–6670. [Google Scholar] [CrossRef]
- Green, M.; Dunlop, E.; Hohl-Ebinger, J.; Yoshita, M.; Kopidakis, N.; Hao, X. Solar Cell Efficiency Tables (Version 57). Prog. Photovolt. Res. Appl. 2021, 29, 3–15. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Xu, J.; Dong, Y.; Chen, B.; Park, S.M.; Ma, D.; Lee, S.; Huang, J.E.; Teale, S. Wide-Bandgap Perovskite Quantum Dots in Perovskite Matrix for Sky-Blue Light-Emitting Diodes. J. Am. Chem. Soc. 2022, 144, 4009–4016. [Google Scholar] [CrossRef]
- Liu, S.; Biju, V.P.; Qi, Y.; Chen, W.; Liu, Z. Recent Progress in the Development of High-Efficiency Inverted Perovskite Solar Cells. NPG Asia Mater. 2023, 15, 27. [Google Scholar] [CrossRef]
- Suresh Kumar, N.; Chandra Babu Naidu, K. A Review on Perovskite Solar Cells (PSCs), Materials and Applications. J. Mater. 2021, 7, 940–956. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Guo, X.; Lu, C.; Wei, J.; Fang, J. Constructing Heterojunctions by Surface Sulfidation for Efficient Inverted Perovskite Solar Cells. Science 2022, 375, 434–437. [Google Scholar] [CrossRef]
- Chen, H.; Teale, S.; Chen, B.; Hou, Y.; Grater, L.; Zhu, T.; Bertens, K.; Park, S.M.; Atapattu, H.R.; Gao, Y.; et al. Quantum-Size-Tuned Heterostructures Enable Efficient and Stable Inverted Perovskite Solar Cells. Nat. Photon. 2022, 16, 352–358. [Google Scholar] [CrossRef]
- Jeng, J.-Y.; Chiang, Y.-F.; Lee, M.-H.; Peng, S.-R.; Guo, T.-F.; Chen, P.; Wen, T.-C. CH3NH3PbI3 Perovskite/Fullerene Planar-Heterojunction Hybrid Solar Cells. Adv. Mater. 2013, 25, 3727–3732. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, Y.; Yue, Y.; Liu, J.; Zhang, W.; Yang, X.; Chen, H.; Bi, E.; Ashraful, I.; Grätzel, M.; et al. Efficient and Stable Large-Area Perovskite Solar Cells with Inorganic Charge Extraction Layers. Science 2015, 350, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Hou, Y.; Bao, C.; Yin, J.; Yuan, F.; Huang, Z.; Song, K.; Liu, J.; Troughton, J.; Gasparini, N.; et al. Managing Grains and Interfaces via Ligand Anchoring Enables 22.3%-Efficiency Inverted Perovskite Solar Cells. Nat. Energy 2020, 5, 131–140. [Google Scholar] [CrossRef]
- Li, Z.; Li, B.; Wu, X.; Sheppard, S.A.; Zhang, S.; Gao, D.; Long, N.J.; Zhu, Z. Organometallic-Functionalized Interfaces for Highly Efficient Inverted Perovskite Solar Cells. Science 2022, 376, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Tong, J.; Xian, Y.; Kerner, R.A.; Dunfield, S.P.; Xiao, C.; Scheidt, R.A.; Kuciauskas, D.; Wang, X.; Hautzinger, M.P.; et al. Surface Reaction for Efficient and Stable Inverted Perovskite Solar Cells. Nature 2022, 611, 278–283. [Google Scholar] [CrossRef]
- Babayigit, A.; Ethirajan, A.; Muller, M.; Conings, B. Toxicity of Organometal Halide Perovskite Solar Cells. Nat. Mater. 2016, 15, 247–251. [Google Scholar] [CrossRef]
- Zhao, X.-G.; Yang, D.; Ren, J.-C.; Sun, Y.; Xiao, Z.; Zhang, L. Rational Design of Halide Double Perovskites for Optoelectronic Applications. Joule 2018, 2, 1662–1673. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, J.; Yin, W.-J.; Yan, Y. Bandgap Engineering of Stable Lead-Free Oxide Double Perovskites for Photovoltaics. Adv. Mater. 2018, 30, 1705901. [Google Scholar] [CrossRef]
- R, S.K.; Akande, A.; El-Mellouhi, F.; Park, H.; Sanvito, S. Theoretical Investigation of the Structural, Elastic, Electronic, and Dielectric Properties of Alkali-Metal-Based Bismuth Ternary Chalcogenides. Phys. Rev. Mater. 2020, 4, 075401. [Google Scholar] [CrossRef]
- Obada, D.O.; Abolade, S.A.; R., S.K.; Ukpong, A.M.; Akande, A. Ab Initio Calculations of the Properties of Defective CsSnCl3: The Role of Anion-Cation Pair Defect. Solid State Ion. 2023, 399, 116262. [Google Scholar] [CrossRef]
- Shao, S.; Liu, J.; Portale, G.; Fang, H.-H.; Blake, G.R.; ten Brink, G.H.; Koster, L.J.A.; Loi, M.A. Highly Reproducible Sn-Based Hybrid Perovskite Solar Cells with 9% Efficiency. Adv. Energy Mater. 2018, 8, 1702019. [Google Scholar] [CrossRef]
- Hoefler, S.F.; Trimmel, G.; Rath, T. Progress on Lead-Free Metal Halide Perovskites for Photovoltaic Applications: A Review. Monatshefte Chem. 2017, 148, 795–826. [Google Scholar] [CrossRef]
- Sun, P.-P.; Li, Q.-S.; Yang, L.-N.; Li, Z.-S. Theoretical Insights into a Potential Lead-Free Hybrid Perovskite: Substituting Pb2+ with Ge2+. Nanoscale 2016, 8, 1503–1512. [Google Scholar] [CrossRef]
- Subramanian, M.A.; Aravamudan, G.; Rao, G.S. Oxide Pyrochlores—A Review. Prog. Solid State Chem. 1983, 15, 55–143. [Google Scholar] [CrossRef]
- Zhao, X.-G.; Yang, D.; Sun, Y.; Li, T.; Zhang, L.; Yu, L.; Zunger, A. Cu–In Halide Perovskite Solar Absorbers. J. Am. Chem. Soc. 2017, 139, 6718–6725. [Google Scholar] [CrossRef]
- Zhao, X.-G.; Yang, J.-H.; Fu, Y.; Yang, D.; Xu, Q.; Yu, L.; Wei, S.-H.; Zhang, L. Design of Lead-Free Inorganic Halide Perovskites for Solar Cells via Cation-Transmutation. J. Am. Chem. Soc. 2017, 139, 2630–2638. [Google Scholar] [CrossRef]
- Wei, F.; Deng, Z.; Sun, S.; Xie, F.; Kieslich, G.; Evans, D.M.; Carpenter, M.A.; Bristowe, P.D.; Cheetham, A.K. The Synthesis, Structure and Electronic Properties of a Lead-Free Hybrid Inorganic–Organic Double Perovskite (MA)2 KBiCl6 (MA = Methylammonium). Mater. Horiz. 2016, 3, 328–332. [Google Scholar] [CrossRef]
- Deng, Z.; Wei, F.; Sun, S.; Kieslich, G.; Cheetham, A.K.; Bristowe, P.D. Exploring the Properties of Lead-Free Hybrid Double Perovskites Using a Combined Computational-Experimental Approach. J. Mater. Chem. A 2016, 4, 12025–12029. [Google Scholar] [CrossRef]
- Volonakis, G.; Haghighirad, A.A.; Milot, R.L.; Sio, W.H.; Filip, M.R.; Wenger, B.; Johnston, M.B.; Herz, L.M.; Snaith, H.J.; Giustino, F. Cs2InAgCl6: A New Lead-Free Halide Double Perovskite with Direct Band Gap. J. Phys. Chem. Lett. 2017, 8, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Tang, Y. First Principle Comparative Study of Transitional Elements Co, Rh, Ir (III)-Based Double Halide Perovskites. Mater. Today Commun. 2023, 34, 105431. [Google Scholar] [CrossRef]
- Vasala, S.; Karppinen, M. A2B′B″O6 Perovskites: A Review. Prog. Solid State Chem. 2015, 43, 1–36. [Google Scholar] [CrossRef]
- Hossain, A.; Bandyopadhyay, P.; Roy, S. An Overview of Double Perovskites A2B′B″O6 with Small Ions at A Site: Synthesis, Structure and Magnetic Properties. J. Alloys Compd. 2018, 740, 414–427. [Google Scholar] [CrossRef]
- Tian, J.; Xue, Q.; Yao, Q.; Li, N.; Brabec, C.J.; Yip, H.-L. Inorganic Halide Perovskite Solar Cells: Progress and Challenges. Adv. Energy Mater. 2020, 10, 2000183. [Google Scholar] [CrossRef]
- Gao, Y.; Pan, Y.; Zhou, F.; Niu, G.; Yan, C. Lead-Free Halide Perovskites: A Review of the Structure–Property Relationship and Applications in Light Emitting Devices and Radiation Detectors. J. Mater. Chem. A 2021, 9, 11931–11943. [Google Scholar] [CrossRef]
- Travis, W.; Glover, E.N.K.; Bronstein, H.; Scanlon, D.O.; Palgrave, R.G. On the Application of the Tolerance Factor to Inorganic and Hybrid Halide Perovskites: A Revised System. Chem. Sci. 2016, 7, 4548–4556. [Google Scholar] [CrossRef]
- Li, C.; Lu, X.; Ding, W.; Feng, L.; Gao, Y.; Guo, Z. Formability of ABX3 (X = F, Cl, Br, I) Halide Perovskites. Acta Crystallogr. Sect. B 2008, 64, 702–707. [Google Scholar] [CrossRef]
- Xiao, Z.; Yan, Y. Progress in Theoretical Study of Metal Halide Perovskite Solar Cell Materials. Adv. Energy Mater. 2017, 7, 1701136. [Google Scholar] [CrossRef]
- Yin, H.; Xian, Y.; Zhang, Y.; Li, W.; Fan, J. Structurally Stabilizing and Environment Friendly Triggers: Double-Metallic Lead-Free Perovskites. Solar RRL 2019, 3, 1900148. [Google Scholar] [CrossRef]
- Wu, Y.; Li, X.; Zeng, H. Lead-Free Halide Double Perovskites: Structure, Luminescence, and Applications. Small Struct. 2021, 2, 2000071. [Google Scholar] [CrossRef]
- Mahmood, Q.; Ghrib, T.; Rached, A.; Laref, A.; Kamran, M.A. Probing of Mechanical, Optical and Thermoelectric Characteristics of Double Perovskites Cs2GeCl/Br6 by DFT Method. Mater. Sci. Semicond. Process. 2020, 112, 105009. [Google Scholar] [CrossRef]
- Iqbal, S.; Mustafa, G.M.; Asghar, M.; Noor, N.A.; Iqbal, M.W.; Mahmood, A.; Shin, Y.-H. Tuning the Optoelectronic and Thermoelectric Characteristics of Narrow Bandgap Rb2AlInX6(X= Cl, Br, I) Double Perovskites: A DFT Study. Mater. Sci. Semicond. Process. 2022, 143, 106551. [Google Scholar] [CrossRef]
- Mahmood, Q.; Younas, M.; Ashiq, M.G.B.; Ramay, S.M.; Mahmood, A.; Ghaithan, H.M. First Principle Study of Lead-Free Double Perovskites Halides Rb2Pd(Cl/Br)6 for Solar Cells and Renewable Energy Devices: A Quantum DFT. Int. J. Energy Res. 2021, 45, 14995–15004. [Google Scholar] [CrossRef]
- Kibbou, M.; Haman, Z.; Bouziani, I.; Khossossi, N.; Benhouria, Y.; Essaoudi, I.; Ainane, A.; Ahuja, R. Cs2InGaX6 (X=Cl, Br, or I): Emergent Inorganic Halide Double Perovskites with Enhanced Optoelectronic Characteristics. Curr. Appl. Phys. 2021, 21, 50–57. [Google Scholar] [CrossRef]
- Noor, N.A.; Iqbal, M.W.; Zelai, T.; Mahmood, A.; Shaikh, H.M.; Ramay, S.M.; Al-Masry, W. Analysis of Direct Band Gap A2ScInI6 (A=Rb, Cs) Double Perovskite Halides Using DFT Approach for Renewable Energy Devices. J. Mater. Res. Technol. 2021, 13, 2491–2500. [Google Scholar] [CrossRef]
- Chrafih, Y.; Al-Hattab, M.; Rahmani, K. Thermodynamic, Optical, and Morphological Studies of the Cs2AgBiX6 Double Perovskites (X = Cl, Br, and I): Insights from DFT Study. J. Alloys Compd. 2023, 960, 170650. [Google Scholar] [CrossRef]
- Rajeev Kumar, N.; Radhakrishnan, R. Electronic, Optical and Mechanical Properties of Lead-Free Halide Double Perovskites Using First-Principles Density Functional Theory. Mater. Lett. 2018, 227, 289–291. [Google Scholar] [CrossRef]
- Iqbal, M.W.; Manzoor, M.; Gouadria, S.; Asghar, M.; Zainab, M.; Ahamd, N.N.; Aftab, S.; Sharma, R.; Zahid, T. DFT Insights on the Opto-Electronic and Thermoelectric Properties of Double Perovskites K2AgSbX6 (X = Cl, Br) via Halides Substitutions for Solar Cell Applications. Mater. Sci. Eng. B 2023, 290, 116338. [Google Scholar] [CrossRef]
- Talebi, M.; Mokhtari, A.; Soleimanian, V. Ab-Inito Simulation of the Structural, Electronic and Optical Properties for the Vacancy-Ordered Double Perovskites A2TiI6 (A = Cs or NH4); a Time-Dependent Density Functional Theory Study. J. Phys. Chem. Solids 2023, 176, 111262. [Google Scholar] [CrossRef]
- Ud Din, M.; Munir, J.; Jamil, M.; Saeed, M.A.; Ain, Q. Electronic Structure and Optical Response of Double Perovskite Rb2NaCoF6 for Optoelectronic Devices. Phys. B Condens. Matter 2022, 627, 413533. [Google Scholar] [CrossRef]
- VanOrman, Z.A.; Nienhaus, L. Recent Advancements in Halide Perovskite Nanomaterials and Their Optoelectronic Applications. InfoMat 2021, 3, 962–986. [Google Scholar] [CrossRef]
- Menedjhi, A.; Bouarissa, N.; Saib, S.; Boucenna, M.; Mezrag, F. Band Structure and Optical Spectra of Double Perovskite Cs2AgBiBr6 for Solar Cells Performance. Acta Phys. Pol. A 2020, 137, 486. [Google Scholar] [CrossRef]
- Sun, Q.; Yin, W.-J.; Wei, S.-H. Searching for Stable Perovskite Solar Cell Materials Using Materials Genome Techniques and High-Throughput Calculations. J. Mater. Chem. C 2020, 8, 12012–12035. [Google Scholar] [CrossRef]
- Slavney, A.H.; Hu, T.; Lindenberg, A.M.; Karunadasa, H.I. A Bismuth-Halide Double Perovskite with Long Carrier Recombination Lifetime for Photovoltaic Applications. J. Am. Chem. Soc. 2016, 138, 2138–2141. [Google Scholar] [CrossRef] [PubMed]
- Filip, M.R.; Hillman, S.; Haghighirad, A.A.; Snaith, H.J.; Giustino, F. Band Gaps of the Lead-Free Halide Double Perovskites Cs2BiAgCl6 and Cs2BiAgBr6 from Theory and Experiment. J. Phys. Chem. Lett. 2016, 7, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, P.; Wei, S.-H. Band Structure Engineering of Cs2AgBiBr6 Perovskite through Order–Disordered Transition: A First-Principle Study. J. Phys. Chem. Lett. 2018, 9, 31–35. [Google Scholar] [CrossRef]
- Liang, L.; Gao, P. Lead-Free Hybrid Perovskite Absorbers for Viable Application: Can We Eat the Cake and Have It Too? Adv. Sci. 2018, 5, 1700331. [Google Scholar] [CrossRef]
- McClure, E.T.; Ball, M.R.; Windl, W.; Woodward, P.M. Cs2AgBiX6 (X = Br, Cl): New Visible Light Absorbing, Lead-Free Halide Perovskite Semiconductors. Chem. Mater. 2016, 28, 1348–1354. [Google Scholar] [CrossRef]
- Hoye, R.L.Z.; Schulz, P.; Schelhas, L.T.; Holder, A.M.; Stone, K.H.; Perkins, J.D.; Vigil-Fowler, D.; Siol, S.; Scanlon, D.O.; Zakutayev, A.; et al. Perovskite-Inspired Photovoltaic Materials: Toward Best Practices in Materials Characterization and Calculations. Chem. Mater. 2017, 29, 1964–1988. [Google Scholar] [CrossRef]
- Volonakis, G.; Filip, M.R.; Haghighirad, A.A.; Sakai, N.; Wenger, B.; Snaith, H.J.; Giustino, F. Lead-Free Halide Double Perovskites via Heterovalent Substitution of Noble Metals. J. Phys. Chem. Lett. 2016, 7, 1254–1259. [Google Scholar] [CrossRef]
- Haug, H.; Koch, S.W. Quantum Theory of the Optical and Electronic Properties of Semiconductors; World Scientific Publishing Company: Singapore, 2009; Volume 10, p. 7184. [Google Scholar]
- Gao, Y.; Wang, X.; Mi, W. Tunable Electronic Structure and Magnetic Properties of Two-Dimensional g-C3N4/Cr2Ge2Te6 van Der Waals Heterostructures. Comput. Mater. Sci. 2021, 187, 110085. [Google Scholar] [CrossRef]
- Komsa, H.-P.; Krasheninnikov, A.V. Native Defects in Bulk and Monolayer MoS2 from First Principles. Phys. Rev. B 2015, 91, 125304. [Google Scholar] [CrossRef]
- Maqsood, S.; Murtaza, G.; Noor, N.A.; Neffati, R.; Nazir, S.; Laref, A. First-Principle Investigation of Thermoelectric and Optoelectronic Properties of Rb2KScI6 and Cs2KScI6 Double Perovskite for Solar Cell Devices. J. Mater. Res. Technol. 2022, 21, 841–849. [Google Scholar] [CrossRef]
- Radja, K.; Farah, B.L.; Ibrahim, A.; Lamia, D.; Fatima, I.; Nabil, B.; Mohamed, A.; Al-Douri, Y.; Abd El-Rehim, A.F. Investigation of Structural, Magneto-Electronic, Elastic, Mechanical and Thermoelectric Properties of Novel Lead-Free Halide Double Perovskite Cs2AgFeCl6: First-Principles Calcuations. J. Phys. Chem. Solids 2022, 167, 110795. [Google Scholar] [CrossRef]
- Aldaghfag, S.A.; Aziz, A.; Younas, A.; Yaseen, M.; Murtaza, A.; Hegazy, H.H. Investigation of Electronic, Optical and Thermoelectric Features of X2ScAgCl6 (X=K, Na) Double Perovskites for Renewable Energy Applications. J. Solid State Chem. 2022, 312, 123179. [Google Scholar] [CrossRef]
- Kattan, N.A.; Rouf, S.A.; Sfina, N.; mana Al-Anazy, M.; Ullah, H.; Hakamy, A.; Mera, A.; Mahmood, Q.; Amin, M.A. Tuning of Band Gap by Anion Variation of Double Perovskites K2AgInX6 (X=Cl, Br) for Solar Cells and Thermoelectric Applications. J. Solid State Chem. 2023, 319, 123820. [Google Scholar] [CrossRef]
- Shah, S.Z.A.; Niaz, S. Lead-Free Indium-Silver Based Double Perovskites for Thermoelectric Applications: Structural, Electronic and Thermoelectric Properties Using First-Principles Approach. Mater. Today Commun. 2021, 28, 102609. [Google Scholar] [CrossRef]
- Aslam, F.; Ullah, H.; Hassan, M. Theoretical Investigation of Cs2InBiX6 (X=Cl, Br, I) Double Perovskite Halides Using First-Principle Calculations. Mater. Sci. Eng. B 2021, 274, 115456. [Google Scholar] [CrossRef]
- Al-Qaisi, S.; Mushtaq, M.; Alomairy, S.; Vu, T.V.; Rached, H.; Haq, B.U.; Mahmood, Q.; Al-Buriahi, M.S. First-Principles Investigations of Na2CuMCl6 (M= Bi, Sb) Double Perovskite Semiconductors: Materials for Green Technology. Mater. Sci. Semicond. Process. 2022, 150, 106947. [Google Scholar] [CrossRef]
- Hu, D.-Y.; Zhao, X.-H.; Tang, T.-Y.; Lu, L.-M.; Li, L.; Gao, L.-K.; Tang, Y.-L. Revealing Structural, Elastic, Electronic and Optical Properties of Potential Perovskites K2CuBiX6 (X=Br, Cl) Based on First-Principles. J. Solid State Chem. 2022, 310, 123046. [Google Scholar] [CrossRef]
- Zhao, S.; Yamamoto, K.; Iikubo, S.; Hayase, S.; Ma, T. First-Principles Study of Electronic and Optical Properties of Lead-Free Double Perovskites Cs2NaBX6 (B = Sb, Bi; X=Cl, Br, I). J. Phys. Chem. Solids 2018, 117, 117–121. [Google Scholar] [CrossRef]
- Manzoor, M.; Iqbal, M.W.; Noor, N.A.; Ullah, H.; Sharma, R.; Alarfaji, S.S. Exploring the Structural, Electronic, Optical, and Thermoelectric Properties of Potassium-Based Double Perovskites K2AgXI6 (X=Sb, Bi) Compounds: A DFT Study. Mater. Sci. Eng. B 2023, 287, 116122. [Google Scholar] [CrossRef]
- Murtaza, G.; Alshahrani, T.; Khalil, R.A.; Mahmood, Q.; Flemban, T.H.; Althib, H.; Laref, A. Lead Free Double Perovsites Halides X2AgTlCl6 (X=Rb, Cs) for Solar Cells and Renewable Energy Applications. J. Solid State Chem. 2021, 297, 121988. [Google Scholar] [CrossRef]
- Maiti, T.; Saxena, M.; Roy, P. Double Perovskite (Sr2B′B″O6) Oxides for High-Temperature Thermoelectric Power Generation—A Review. J. Mater. Res. 2019, 34, 107–125. [Google Scholar] [CrossRef]
- Bhandari, S.R.; Yadav, D.K.; Belbase, B.P.; Zeeshan, M.; Sadhukhan, B.; Rai, D.P.; Thapa, R.K.; Kaphle, G.C.; Ghimire, M.P. Electronic, Magnetic, Optical and Thermoelectric Properties of Ca2Cr1−xNixOsO6 Double Perovskites. RSC Adv. 2020, 10, 16179–16186. [Google Scholar] [CrossRef] [PubMed]
- Albalawi, H.; Mustafa, G.M.; Saba, S.; Kattan, N.A.; Mahmood, Q.; Somaily, H.H.; Morsi, M.; Alharthi, S.; Amin, M.A. Study of Optical and Thermoelectric Properties of Double Perovskites Cs2KTlX6 (X = Cl, Br, I) for Solar Cell and Energy Harvesting. Mater. Today Commun. 2022, 32, 104083. [Google Scholar] [CrossRef]
- Flemban, T.H.; Zelai, T.; Mahmood, Q.; Devi, A.A.S.; Sajjad, M.; Alhossainy, M.H.; Somaily, H.H.; Mera, A.; Alharthi, S.; Amin, M.A. Half-Metallic Ferromagnetism and Thermoelectric Properties of Double Perovskites Rb2Z(Cl/Br)6 (Z = Ta, W, Re). J. Alloys Compd. 2022, 894, 162313. [Google Scholar] [CrossRef]
- Khandy, S.A.; Gupta, D.C. Electronic Structure, Magnetism and Thermoelectric Properties of Double Perovskite Sr2HoNbO6. J. Magn. Magn. Mater. 2018, 458, 176–182. [Google Scholar] [CrossRef]
- Saxena, M.; Maiti, T. Effect of Ba-Doping on High Temperature Thermoelectric Properties of Sr2TiMoO6 Double Perovskites. J. Alloys Compd. 2017, 710, 472–478. [Google Scholar] [CrossRef]
- Sudha; Saxena, M.; Balani, K.; Maiti, T. Structure and Thermoelectric Properties of Calcium Doped Sr2TiCoO6 Double Perovskites. Mater. Sci. Eng. B 2019, 244, 65–71. [Google Scholar] [CrossRef]
- Guechi, N.; Bouhemadou, A.; Bin-Omran, S.; Bourzami, A.; Louail, L. Elastic, Optoelectronic and Thermoelectric Properties of the Lead-Free Halide Semiconductors Cs2AgBiX6 (X=Cl, Br): Ab Initio Investigation. J. Electron. Mater. 2018, 47, 1533–1545. [Google Scholar] [CrossRef]
- Senkov, O.N.; Miracle, D.B. Generalization of Intrinsic Ductile-to-Brittle Criteria by Pugh and Pettifor for Materials with a Cubic Crystal Structure. Sci. Rep. 2021, 11, 4531. [Google Scholar] [CrossRef]
- Greaves, G.N.; Greer, A.L.; Lakes, R.S.; Rouxel, T. Poisson’s Ratio and Modern Materials. Nat. Mater. 2011, 10, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Anbarasan, R.; Balasubramani, V.; Srinivasan, M.; Sundar, J.K.; Ramasamy, P.; Al-Kahtani, A.A.; Ubaidullah, M.; Setiawan, I.A.; Kim, W.K.; Gedi, S. First Principle Insights on Mechanical, Electronic and Optical Properties of Direct Bandgap Material Cs2KScX6 (X=Cl, Br and I) for Optoelectronic Applications. J. Solid State Chem. 2022, 316, 123590. [Google Scholar] [CrossRef]
- Gubernatis, J.E.; Lookman, T. Machine Learning in Materials Design and Discovery: Examples from the Present and Suggestions for the Future. Phys. Rev. Mater. 2018, 2, 120301. [Google Scholar] [CrossRef]
- Jacobs, R.; Luo, G.; Morgan, D. Materials Discovery of Stable and Nontoxic Halide Perovskite Materials for High-Efficiency Solar Cells. Adv. Funct. Mater. 2019, 29, 1804354. [Google Scholar] [CrossRef]
- Allam, O.; Holmes, C.; Greenberg, Z.; Kim, K.C.; Jang, S.S. Density Functional Theory—Machine Learning Approach to Analyze the Bandgap of Elemental Halide Perovskites and Ruddlesden-Popper Phases. ChemPhysChem 2018, 19, 2559–2565. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, J.; Wang, Z.; Lu, X.-G.; Avdeev, M.; Shi, S.; Wang, C.; Yu, T. Predicting Creep Rupture Life of Ni-Based Single Crystal Superalloys Using Divide-and-Conquer Approach Based Machine Learning. Acta Mater. 2020, 195, 454–467. [Google Scholar] [CrossRef]
- Pilania, G.; Mannodi-Kanakkithodi, A.; Uberuaga, B.P.; Ramprasad, R.; Gubernatis, J.E.; Lookman, T. Machine Learning Bandgaps of Double Perovskites. Sci. Rep. 2016, 6, 19375. [Google Scholar] [CrossRef] [PubMed]
- Ramprasad, R.; Batra, R.; Pilania, G.; Mannodi-Kanakkithodi, A.; Kim, C. Machine Learning in Materials Informatics: Recent Applications and Prospects. NPJ Comput. Mater. 2017, 3, 54. [Google Scholar] [CrossRef]
- Himanen, L.; Geurts, A.; Foster, A.S.; Rinke, P. Data-Driven Materials Science: Status, Challenges, and Perspectives. Adv. Sci. 2019, 6, 1900808. [Google Scholar] [CrossRef]
- Sun, J.-P.; McKeown Wessler, G.C.; Wang, T.; Zhu, T.; Blum, V.; Mitzi, D.B. Structural Tolerance Factor Approach to Defect-Resistant I2-II-IV-X4 Semiconductor Design. Chem. Mater. 2020, 32, 1636–1649. [Google Scholar] [CrossRef]
- Wan, X.; Feng, W.; Wang, Y.; Wang, H.; Zhang, X.; Deng, C.; Yang, N. Materials Discovery and Properties Prediction in Thermal Transport via Materials Informatics: A Mini Review. Nano Lett. 2019, 19, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Shafizadeh, A.; Shahbeig, H.; Nadian, M.H.; Mobli, H.; Dowlati, M.; Gupta, V.K.; Peng, W.; Lam, S.S.; Tabatabaei, M.; Aghbashlo, M. Machine Learning Predicts and Optimizes Hydrothermal Liquefaction of Biomass. Chem. Eng. J. 2022, 445, 136579. [Google Scholar] [CrossRef]
- Toyao, T.; Maeno, Z.; Takakusagi, S.; Kamachi, T.; Takigawa, I.; Shimizu, K. Machine Learning for Catalysis Informatics: Recent Applications and Prospects. ACS Catal. 2020, 10, 2260–2297. [Google Scholar] [CrossRef]
- Chen, C.; Zuo, Y.; Ye, W.; Li, X.; Deng, Z.; Ong, S.P. A Critical Review of Machine Learning of Energy Materials. Adv. Energy Mater. 2020, 10, 1903242. [Google Scholar] [CrossRef]
- Zhou, T.; Song, Z.; Sundmacher, K. Big Data Creates New Opportunities for Materials Research: A Review on Methods and Applications of Machine Learning for Materials Design. Engineering 2019, 5, 1017–1026. [Google Scholar] [CrossRef]
- Shi, L.; Chang, D.; Ji, X.; Lu, W. Using Data Mining To Search for Perovskite Materials with Higher Specific Surface Area. J. Chem. Inf. Model. 2018, 58, 2420–2427. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Xu, P.; Li, M.; Lu, W. Machine Learning for Perovskite Materials Design and Discovery. NPJ Comput. Mater. 2021, 7, 23. [Google Scholar] [CrossRef]
- Talapatra, A.; Uberuaga, B.P.; Stanek, C.R.; Pilania, G. Band Gap Predictions of Double Perovskite Oxides Using Machine Learning. Commun. Mater. 2023, 4, 46. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Zhou, X. Machine-Learning Prediction of the Computed Band Gaps of Double Perovskite Materials. arXiv 2023, arXiv:2301.03372. [Google Scholar]
- Obada, D.O.; Okafor, E.; Abolade, S.A.; Ukpong, A.M.; Dodoo-Arhin, D.; Akande, A. Explainable Machine Learning for Predicting the Band Gaps of ABX3 Perovskites. Mater. Sci. Semicond. Process. 2023, 161, 107427. [Google Scholar] [CrossRef]
- Kirklin, S.; Saal, J.E.; Meredig, B.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C. The Open Quantum Materials Database (OQMD): Assessing the Accuracy of DFT Formation Energies. NPJ Comput. Mater. 2015, 1, 15010. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G. Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Landis, D.D.; Hummelshøj, J.S.; Nestorov, S.; Greeley, J.; Dułak, M.; Bligaard, T.; Nørskov, J.K.; Jacobsen, K.W. The Computational Materials Repository. Comput. Sci. Eng. 2012, 14, 51–57. [Google Scholar] [CrossRef]
- Curtarolo, S. Using Your Own Computer to Search for Novel Materials (with a Little Help from the Aflowlib. Org Consortium Online Library). Bull. Am. Phys. Soc. 2014, 59, 273. [Google Scholar]
- Curtarolo, S.; Setyawan, W.; Wang, S.; Xue, J.; Yang, K.; Taylor, R.H.; Nelson, L.J.; Hart, G.L.; Sanvito, S.; Buongiorno-Nardelli, M. AFLOWLIB. ORG: A Distributed Materials Properties Repository from High-Throughput Ab Initio Calculations. Comput. Mater. Sci. 2012, 58, 227–235. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obada, D.O.; Akinpelu, S.B.; Abolade, S.A.; Okafor, E.; Ukpong, A.M.; Kumar R, S.; Akande, A. Lead-Free Double Perovskites: A Review of the Structural, Optoelectronic, Mechanical, and Thermoelectric Properties Derived from First-Principles Calculations, and Materials Design Applicable for Pedagogical Purposes. Crystals 2024, 14, 86. https://doi.org/10.3390/cryst14010086
Obada DO, Akinpelu SB, Abolade SA, Okafor E, Ukpong AM, Kumar R S, Akande A. Lead-Free Double Perovskites: A Review of the Structural, Optoelectronic, Mechanical, and Thermoelectric Properties Derived from First-Principles Calculations, and Materials Design Applicable for Pedagogical Purposes. Crystals. 2024; 14(1):86. https://doi.org/10.3390/cryst14010086
Chicago/Turabian StyleObada, David O., Shittu B. Akinpelu, Simeon A. Abolade, Emmanuel Okafor, Aniekan M. Ukpong, Syam Kumar R, and Akinlolu Akande. 2024. "Lead-Free Double Perovskites: A Review of the Structural, Optoelectronic, Mechanical, and Thermoelectric Properties Derived from First-Principles Calculations, and Materials Design Applicable for Pedagogical Purposes" Crystals 14, no. 1: 86. https://doi.org/10.3390/cryst14010086
APA StyleObada, D. O., Akinpelu, S. B., Abolade, S. A., Okafor, E., Ukpong, A. M., Kumar R, S., & Akande, A. (2024). Lead-Free Double Perovskites: A Review of the Structural, Optoelectronic, Mechanical, and Thermoelectric Properties Derived from First-Principles Calculations, and Materials Design Applicable for Pedagogical Purposes. Crystals, 14(1), 86. https://doi.org/10.3390/cryst14010086