
Graphene-Like ZnO: A Mini Review



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
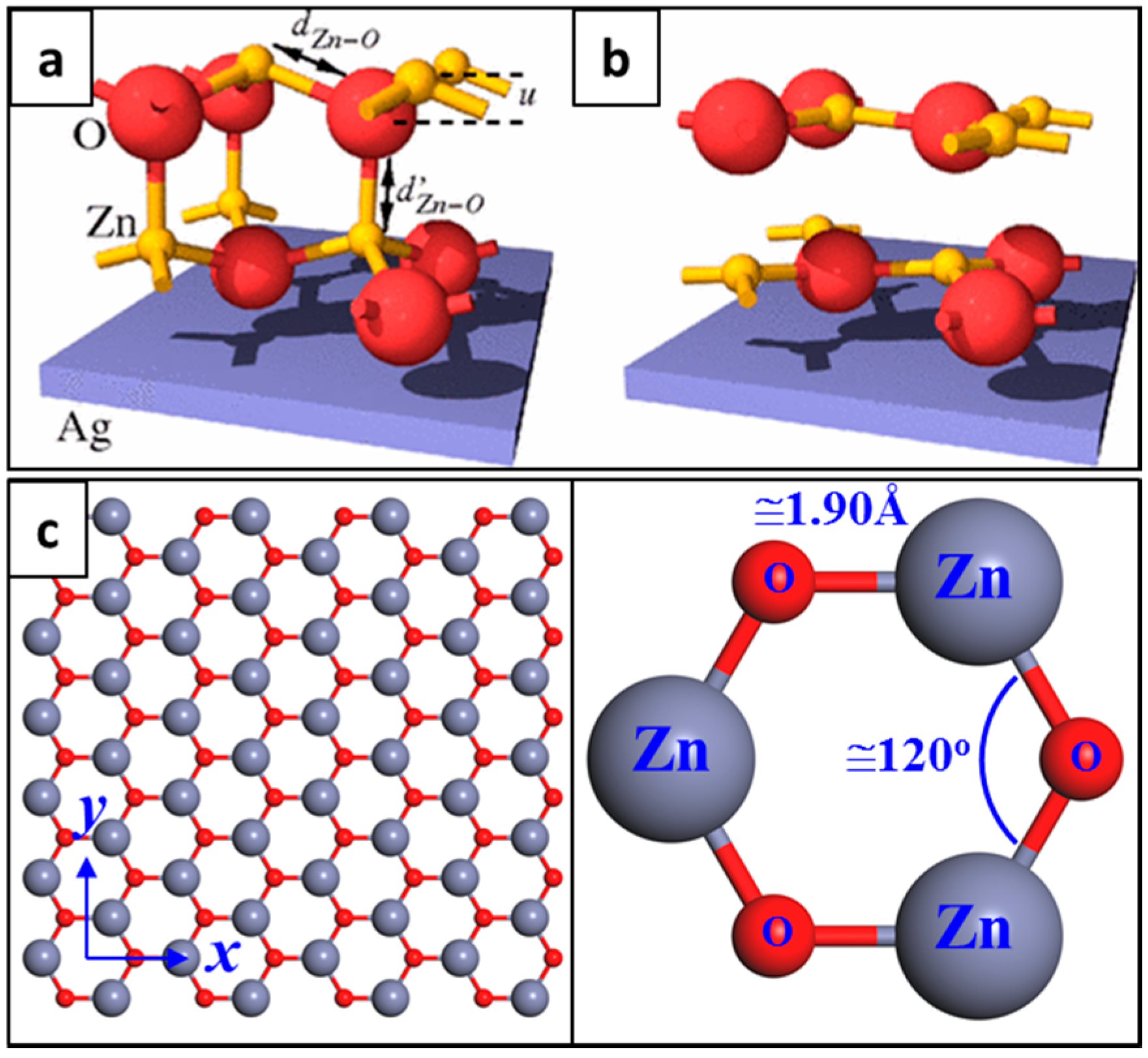
2. Structure of Graphene-Like ZnO (g-ZnO)
3. Properties of Graphene-Like ZnO
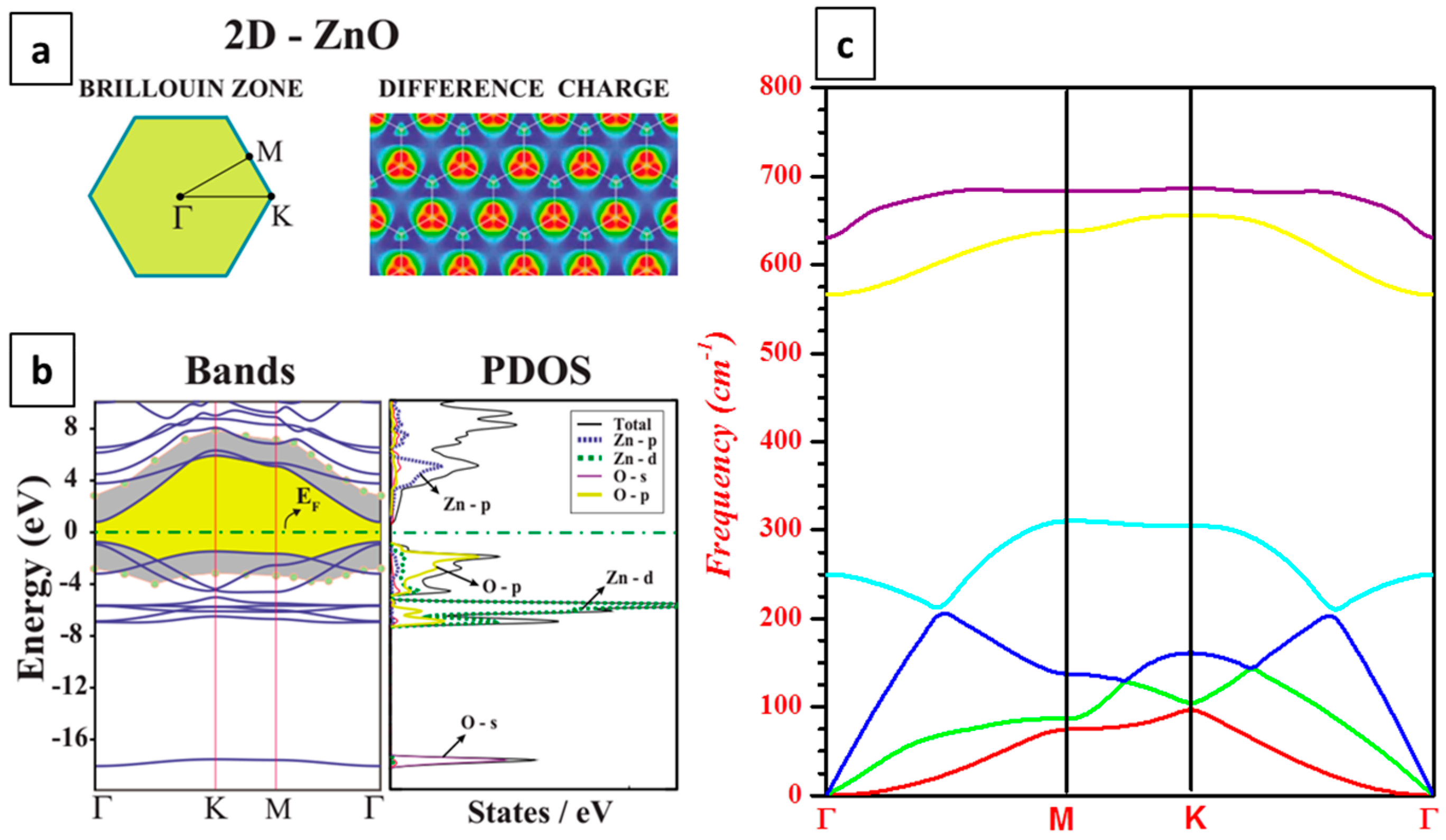
3.1. Electronic Structure
3.2. Phonon Dispersion Relations
3.3. Mechanical Properties
3.4. Elastic and Piezoelectric Properties
4. Synthesis of Graphene-Like ZnO
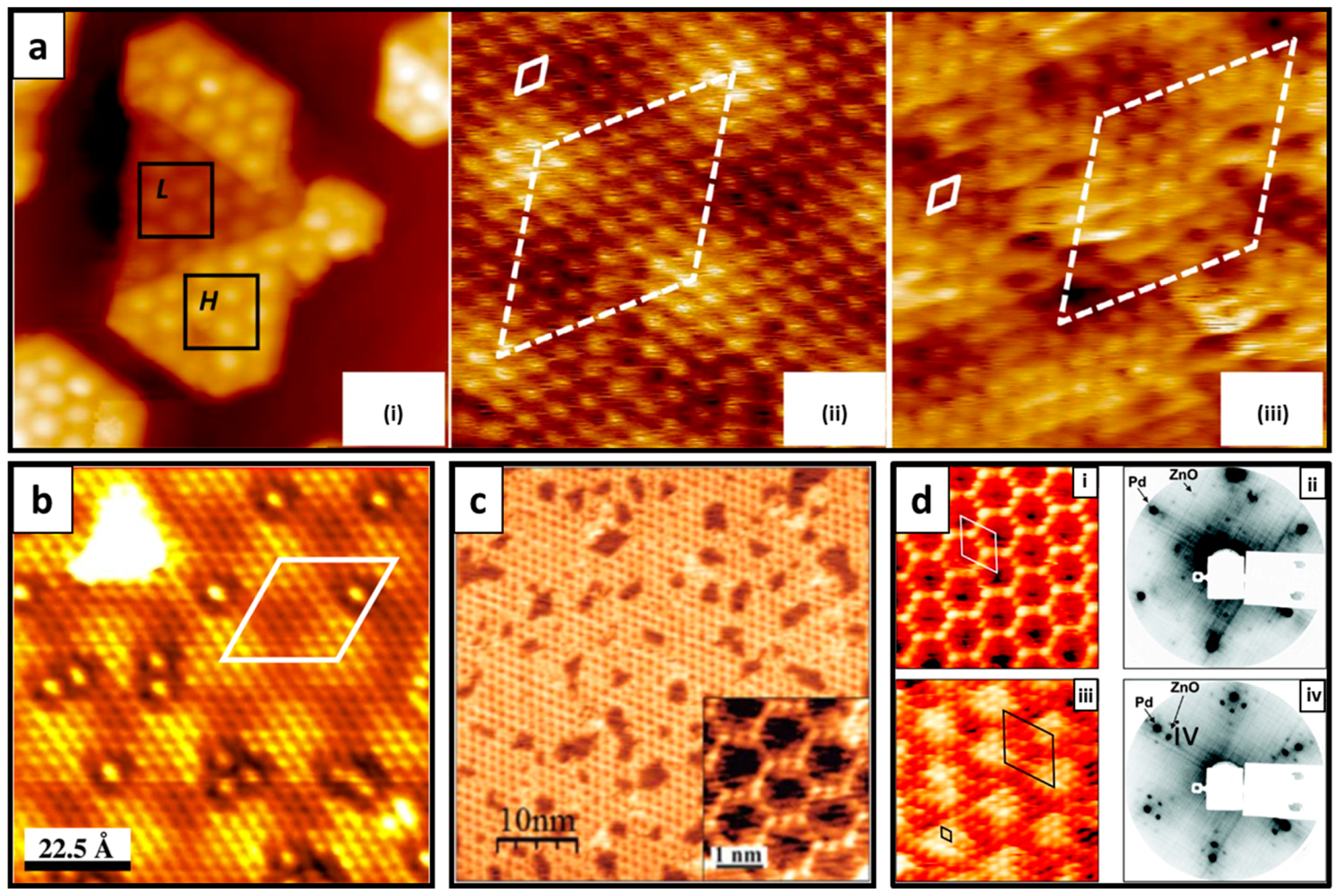
4.1. g-ZnO Growth over a Metal Substrate
4.2. Hydrothermal Growth
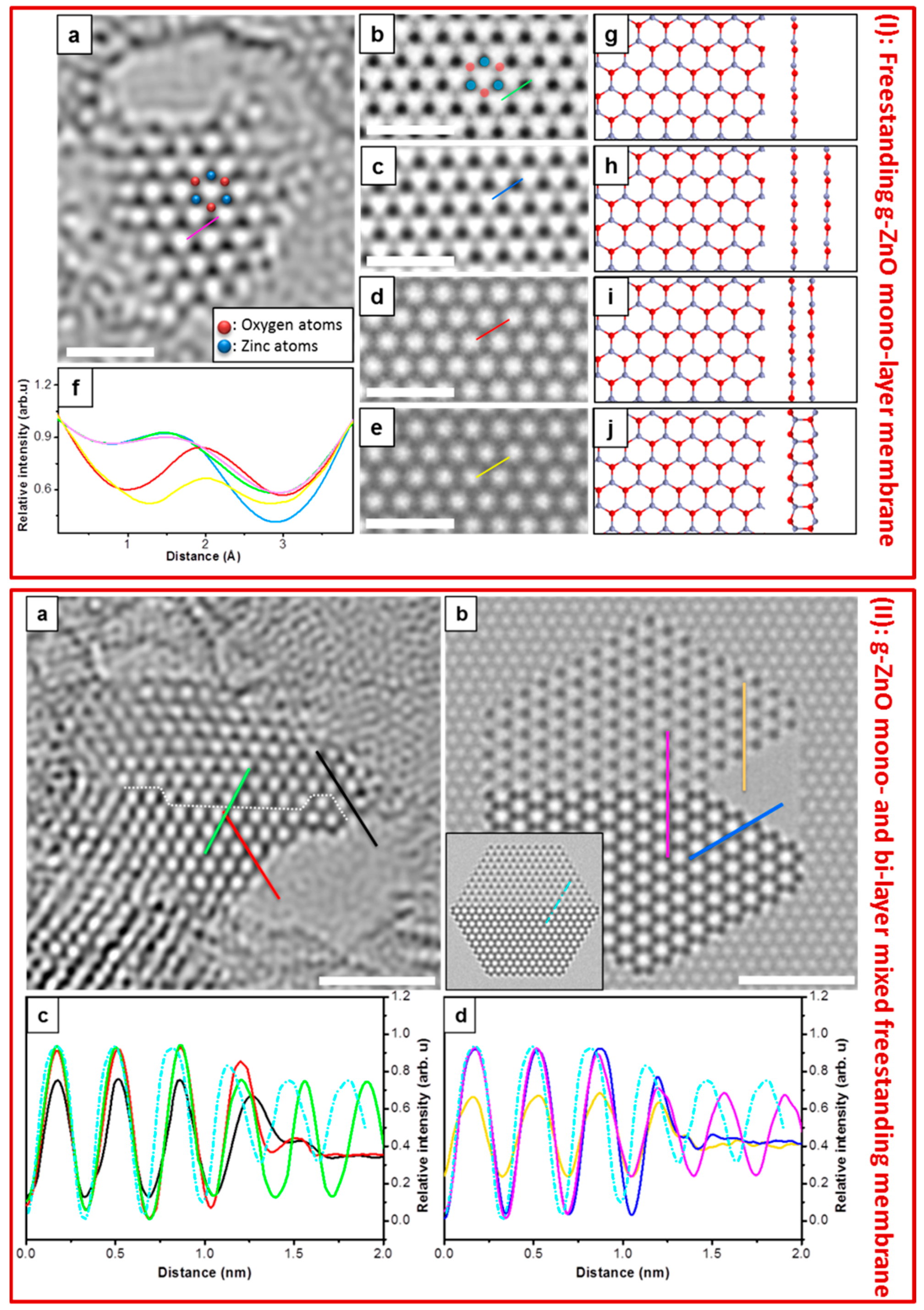
4.3. Free-Standing Formation of g-ZnO in Graphene Pores
5. Modification and Potential Applications of g-ZnO
5.1. g-ZnO Structure Modification
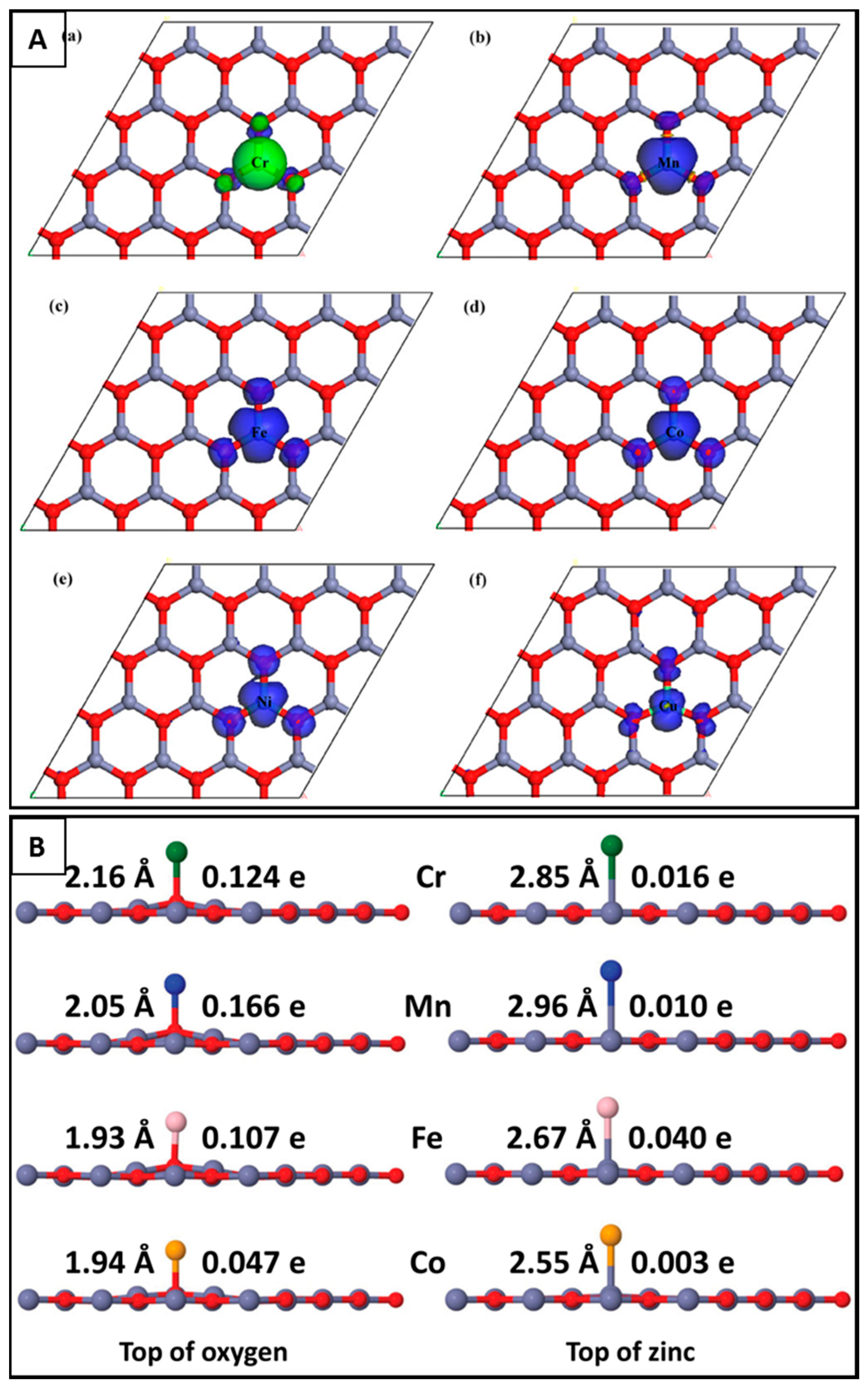
5.1.1. Doping and Decorating of g-ZnO
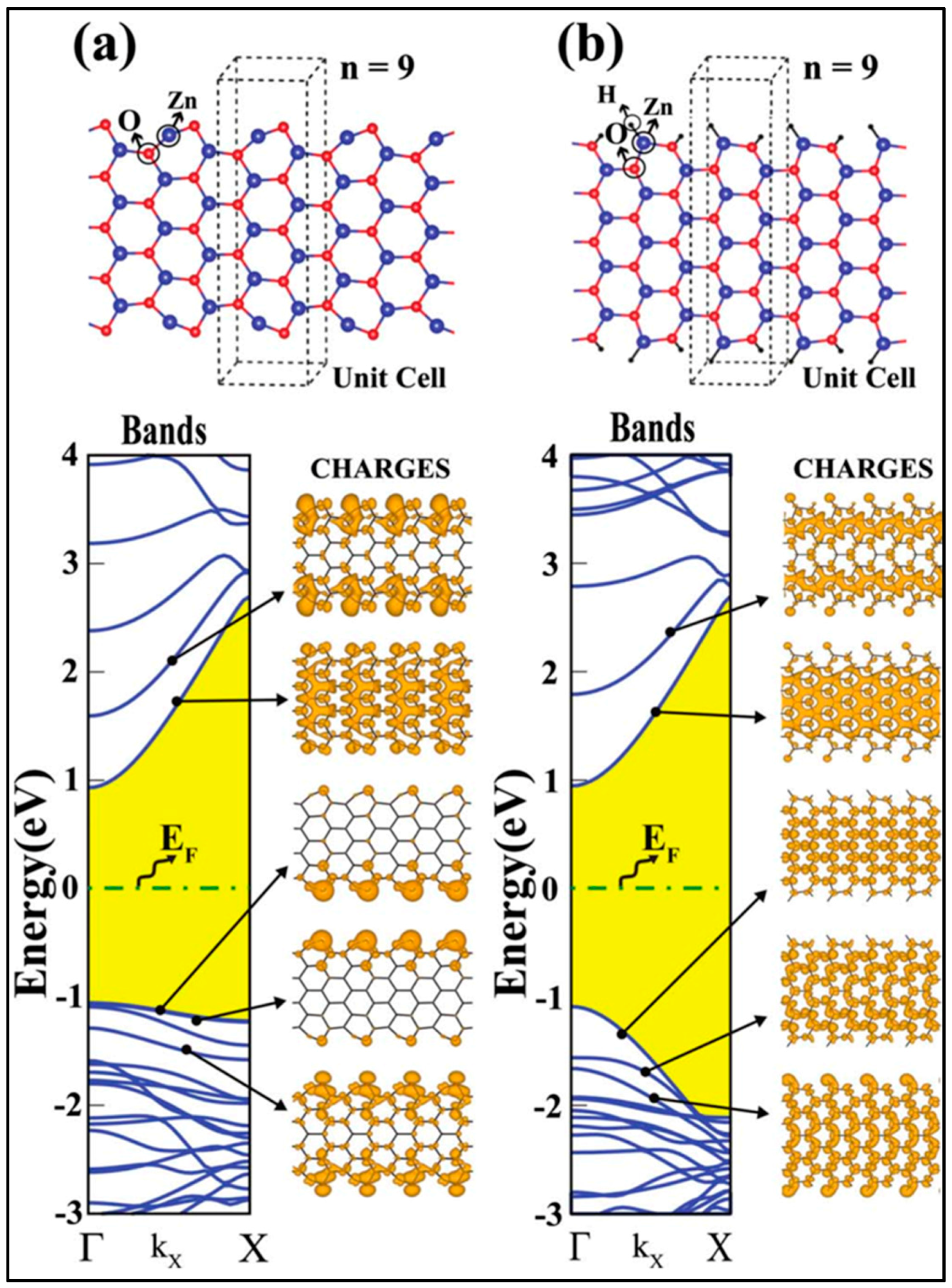
5.1.2. g-ZnO Nanoribbon
5.2. Potential Applications
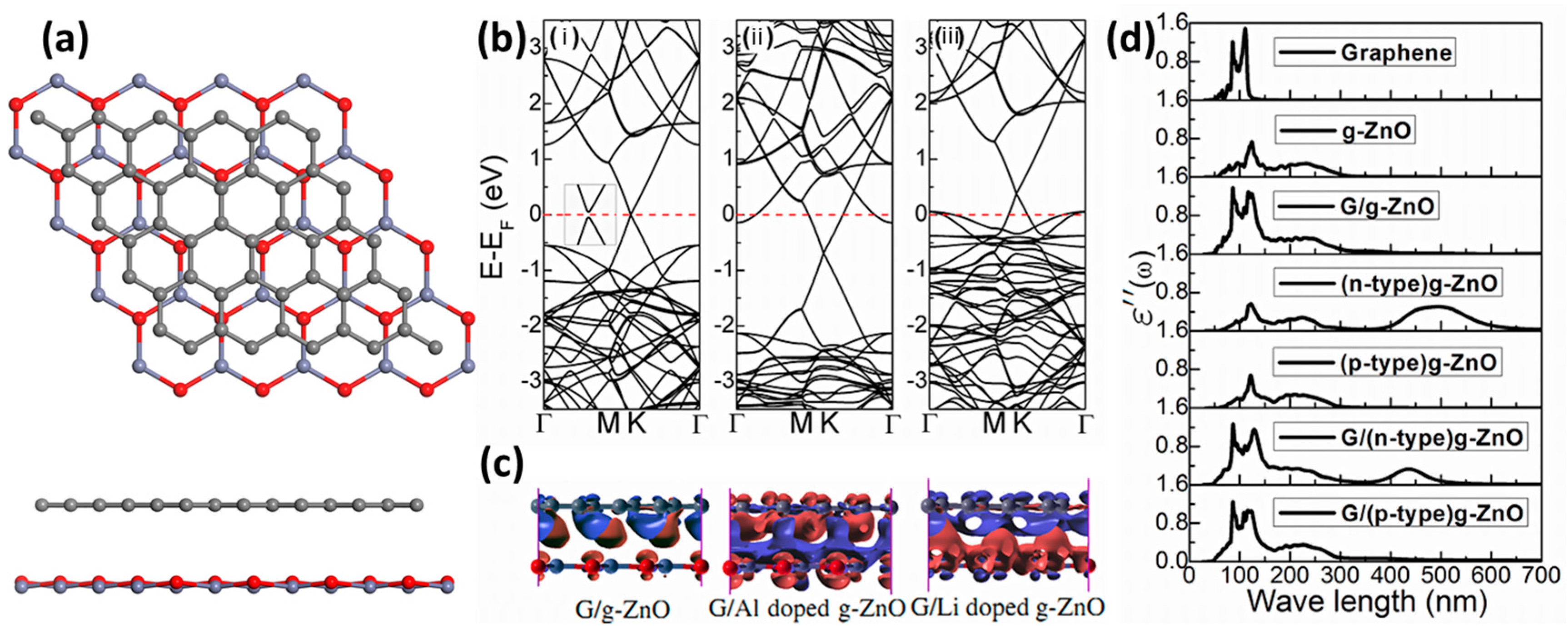
5.2.1. Graphene–g-ZnO Heterostructures
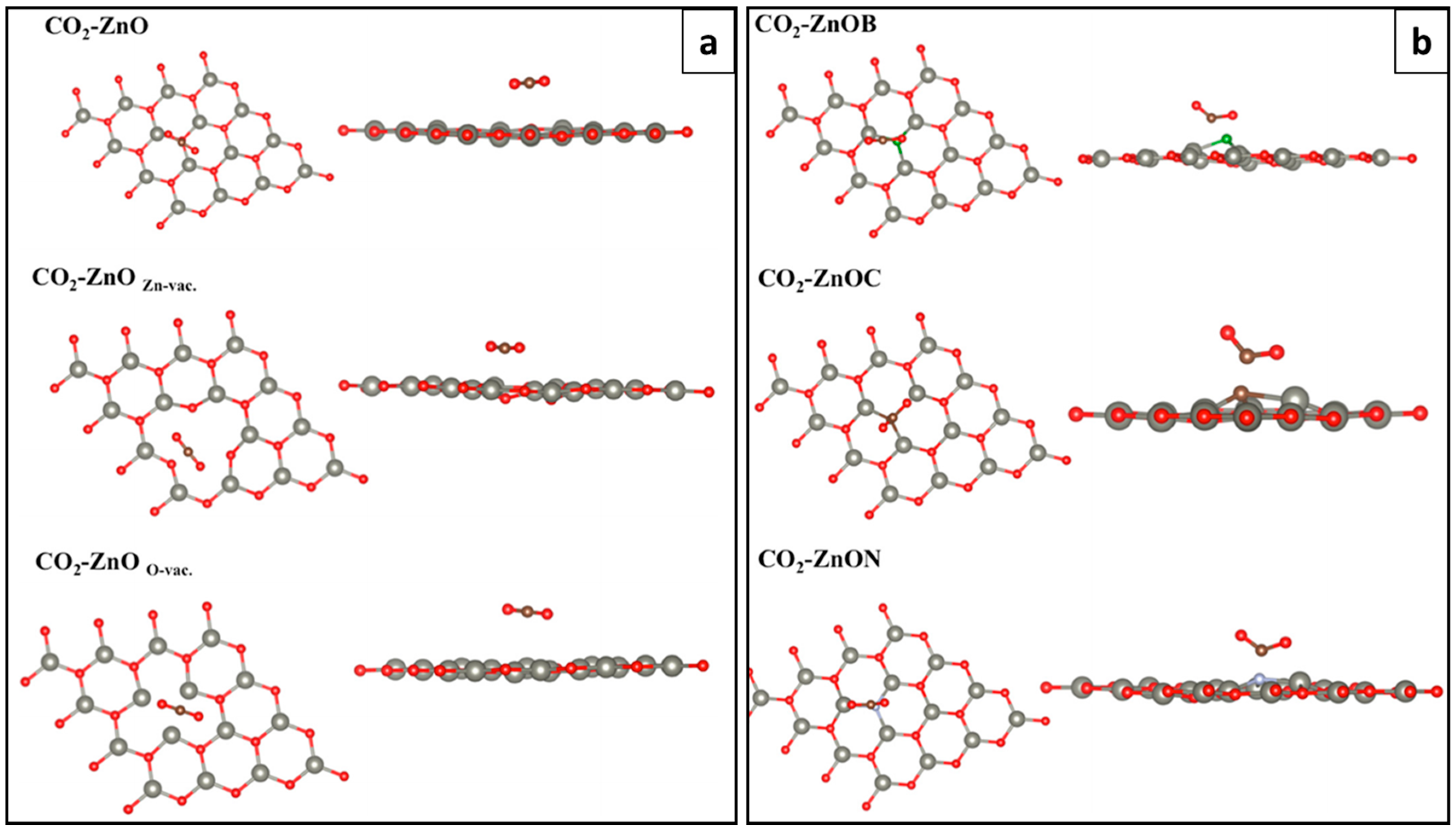
5.2.2. CO and CO2 Physisorption of g-ZnO
5.2.3. Catalytic Activity g-ZnO
6. Summary and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, Z.L. Zinc oxide nanostructures: Growth, properties and applications. J. Phys. Condens. Matter 2004, 16, 829–858. [Google Scholar] [CrossRef]
- Fan, Z.; Lu, J.G. Zinc oxide nanostructures: Synthesis and properties. J Nanosci. Nanotechnol. 2005, 5, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Claeyssens, F.; Freeman, C.L.; Allan, N.L.; Sun, Y.; Ashfolda, M.N.R.; Harding, J.H. Growth of ZnO thin films-experiment and theory. J. Mater. Chem. 2005, 15, 139–148. [Google Scholar] [CrossRef]
- Tusche, C.; Meyerheim, H.L.; Kirschner, J. Observation of depolarized ZnO(0001) mono-layers: Formation of unreconstructed planar sheets. Phys. Rev. Lett. 2007, 99, 026102. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.C.; Hu, X. Elasticity and piezoelectricity of zinc oxide crystals, single layers, and possible single-walled nanotubes. Phys. Rev. B 2006, 74, 035434. [Google Scholar] [CrossRef]
- Topsakal, M.; Cahangirov, S.; Bekaroglu, E.; Ciraci, S. First-principles study of zinc oxide honeycomb structures. Phys. Rev. B 2009, 80, 235119. [Google Scholar] [CrossRef]
- Guo, H.; Zhao, Y.; Lu, N.; Kan, E.; Zeng, X.C.; Wu, X.; Yang, J. Tunable magnetism in a nonmetal-substituted ZnO mono-layer: A first-principles study. J. Phys. Chem. C 2012, 116, 11336–11342. [Google Scholar] [CrossRef]
- Zheng, F.; Zhang, C.; Wang, P.; Luan, H. Tuning the electronic and magnetic properties of carbon-doped ZnO nanosheets: First-principles prediction. J. Appl. Phys. 2012, 111, 044329. [Google Scholar] [CrossRef]
- Li, C.; Guo, W.; Kong, Y.; Gao, H. First-principles study on ZnO nanoclusters with hexagonal prism structures. Appl. Phys. Lett. 2007, 90, 223102. [Google Scholar] [CrossRef]
- Tu, Z.C. First-principles study on physical properties of a single ZnO mono-layer with graphene-like structure. J. Comput. Theor. Nanos. 2010, 7, 1182–1186. [Google Scholar] [CrossRef]
- Chai, G.L.; Lin, C.S.; Cheng, W.D. First-principles study of ZnO cluster-decorated carbon nanotubes. Nanotechnology 2011, 22, 445705. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.K.; Yadav, P.S.; Agrawal, S.; Agrawal, B.K. Structural and electronic properties of ZnO nanoclusters: A B3LYP DFT study. Adv. Mater. Res. 2013, 650, 29–33. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Eklund, P.C. Phonons in Carbon Nanotubes. Adv. Phys. 2000, 49, 705–814. [Google Scholar] [CrossRef]
- Peng, Q.; Liang, C.; Ji, W.; De, S. A first principles investigation of the mechanical properties of g-ZnO: The graphene-like hexagonal zinc oxide mono-layer. Comp. Mater. Sci. 2013, 68, 320–324. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals; Clarendon Press: Oxford, UK, 1985; pp. 110–148. [Google Scholar]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Clarendon Press: Oxford, UK, 1954. [Google Scholar]
- Weirum, G.; Barcaro, G.; Fortunelli, A.; Weber, F.; Schennach, R.; Surnev, S.; Netzer, F.P. Growth and surface structure of zinc oxide layers on a Pd(111) surface. J. Phys. Chem. C 2010, 114, 15432–15439. [Google Scholar] [CrossRef]
- Liu, B.H.; McBriarty, M.E.; Bedzyk, M.J.; Shaikhutdinov, S.; Freund, H.J. Structural transformations of zinc oxide layers on Pt(111). J. Phys. Chem. C 2014, 118, 28725–28729. [Google Scholar] [CrossRef]
- Liu, B.H.; Boscoboinik, A.J.; Cui, Y.; Shaikhutdinov, S.; Freund, H.J. Stabilization of ultrathin zinc oxide films on metals: Reconstruction versus hydroxylation. J. Phys. Chem. C 2015, 119, 7842–7847. [Google Scholar] [CrossRef]
- Deng, X.; Yao, K.; Sun, K.; Li, W.X.; Lee, J.; Matranga, C. Growth of single- and bi-layer ZnO on Au(111) and interaction with copper. J. Phys. Chem. C 2013, 117, 11211–11218. [Google Scholar] [CrossRef]
- Shiotari, A.; Liu, B.H.; Jaekel, S.; Grill, L.; Shaikhutdinov, S.; Freund, H.J.; Wolf, M.; Kumagai, T. Local characterization of ultrathin ZnO layers on Ag(111) by scanning tunneling microscopy and atomic force microscopy. J. Phys. Chem. C 2014, 118, 27428–27435. [Google Scholar] [CrossRef]
- Pan, Q.; Liu, B.H.; McBriarty, M.E.; Martynova, Y.; Groot, I.M.N.; Wang, S.; Bedzyk, M.J.; Shaikhutdinov, S.; Freund, H.J. Reactivity of ultra-thin ZnO films supported by Ag(111) and Cu(111): A comparison to ZnO/Pt(111). Catal. Lett. 2014, 144, 648–655. [Google Scholar] [CrossRef]
- Schott, V.; Oberhofer, H.; Birkner, A.; Xu, M.; Wang, Y.; Muhler, M.; Reuter, K.; Woll, C. Chemical activity of thin oxide layers: Strong interactions with the support yield a new thin-film phase of ZnO. Angew. Chem. Int. Ed. 2013, 52, 11925–11929. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; Nayak, S.K.; Chelliah, P.; Rath, M.K.; Paridac, B. Observations of two-dimensional mono-layer zinc oxide. Mater. Res. Bull. 2016, 75, 134–138. [Google Scholar] [CrossRef]
- Ta, H.Q.; Bachmatiuk, A.; Dianat, A.; Ortmann, F.; Zhao, J.; Warner, J.H.; Eckert, J.; Cunniberti, G.; Rümmeli, M.H. In-situ observations of freestanding graphene-like mono- and bi-layer ZnO membranes. ACS Nano 2015, 9, 11408–11413. [Google Scholar]
- Schmidt, T.M.; Miwa, R.H.; Fazzio, A. Ferromagnetic coupling in a Co-doped graphenelike ZnO sheet. Phys. Rev. B 2010, 81, 195413. [Google Scholar] [CrossRef]
- He, A.L.; Wang, X.Q.; Wu, R.Q.; Lu, Y.H.; Feng, Y.P. Adsorption of an Mn atom on a ZnO sheet and nanotube: A density functional theory study. J. Phys. Condens. Matter 2010, 22, 175501. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, H.; Cheng, X. Electronic and magnetic properties of all 3D transition-metal-doped ZnO mono-layers. Int. J. Quantum Chem. 2013, 113, 2243–2250. [Google Scholar] [CrossRef]
- Lei, J.; Xu, M.C.; Hu, S.J. Transition metal decorated graphene-like zinc oxide mono-layer: A first-principles investigation. J. Appl. Phys. 2015, 118, 104302. [Google Scholar] [CrossRef]
- Botello-Méndez, A.R.; Martínez-Martínez, M.T.; López-Urías, F.; Terrones, M.; Terrones, H. Metallic edges in zinc oxide nanoribbons. Chem. Phys. Lett. 2007, 448, 258–263. [Google Scholar] [CrossRef]
- Botello-Méndez, A.R.; López-Urías, F.; Terrones, M.; Terrones, H. Magnetic behavior in zinc oxide zigzag nanoribbon. Nano Lett. 2008, 8, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Li, Z.; Yang, J. Electronic and optical properties of graphene and graphitic ZnO nanocomposite structures. J. Chem. Phys. 2013, 138, 124706. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Liu, Y.; Lu, R.; Xiao, C.; Deng, K.; Kan, E. Will a graphitic-like ZnO single-layer be an ideal substrate for graphene? RSC Adv. 2014, 4, 17478. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Zhang, M.L.; Zhou, Y.C.; Zhao, J.H.; Fanga, S.M.; Li, F. Tunable electronic and magnetic properties of graphene-like ZnO mono-layer upon doping and CO adsorption: A first-principles study. J. Mater. Chem. A 2014, 2, 13129. [Google Scholar] [CrossRef]
- Rao, G.S.; Hussain, T.; Islam, M.S.; Sagynbaeva, M.; Gupta, D.; Panigrahi, P.; Ahuja, R. Adsorption mechanism of graphene-like ZnO mono-layer towards CO2 molecules: enhanced CO2 capture. Nanotechnology 2016, 27, 015502. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wang, Q.; Li, T.; Tang, Z.; Yang, G.; He, C.; Lu, Z. CO catalytic oxidation on Al-doped graphene-like ZnO mono-layer sheets: A first-principles study. J. Mater. Chem. C 2015, 3, 9964–9972. [Google Scholar] [CrossRef]









© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ta, H.Q.; Zhao, L.; Pohl, D.; Pang, J.; Trzebicka, B.; Rellinghaus, B.; Pribat, D.; Gemming, T.; Liu, Z.; Bachmatiuk, A.; et al. Graphene-Like ZnO: A Mini Review. Crystals 2016, 6, 100. https://doi.org/10.3390/cryst6080100
Ta HQ, Zhao L, Pohl D, Pang J, Trzebicka B, Rellinghaus B, Pribat D, Gemming T, Liu Z, Bachmatiuk A, et al. Graphene-Like ZnO: A Mini Review. Crystals. 2016; 6(8):100. https://doi.org/10.3390/cryst6080100
Chicago/Turabian StyleTa, Huy Q., Liang Zhao, Darius Pohl, Jinbo Pang, Barbara Trzebicka, Bernd Rellinghaus, Didier Pribat, Thomas Gemming, Zhongfan Liu, Alicja Bachmatiuk, and et al. 2016. "Graphene-Like ZnO: A Mini Review" Crystals 6, no. 8: 100. https://doi.org/10.3390/cryst6080100
APA StyleTa, H. Q., Zhao, L., Pohl, D., Pang, J., Trzebicka, B., Rellinghaus, B., Pribat, D., Gemming, T., Liu, Z., Bachmatiuk, A., & Rümmeli, M. H. (2016). Graphene-Like ZnO: A Mini Review. Crystals, 6(8), 100. https://doi.org/10.3390/cryst6080100