
Structural Characterization of Febuxostat/l-Pyroglutamic Acid Cocrystal Using Solid-State 13C-NMR and Investigational Study of Its Water Solubility

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
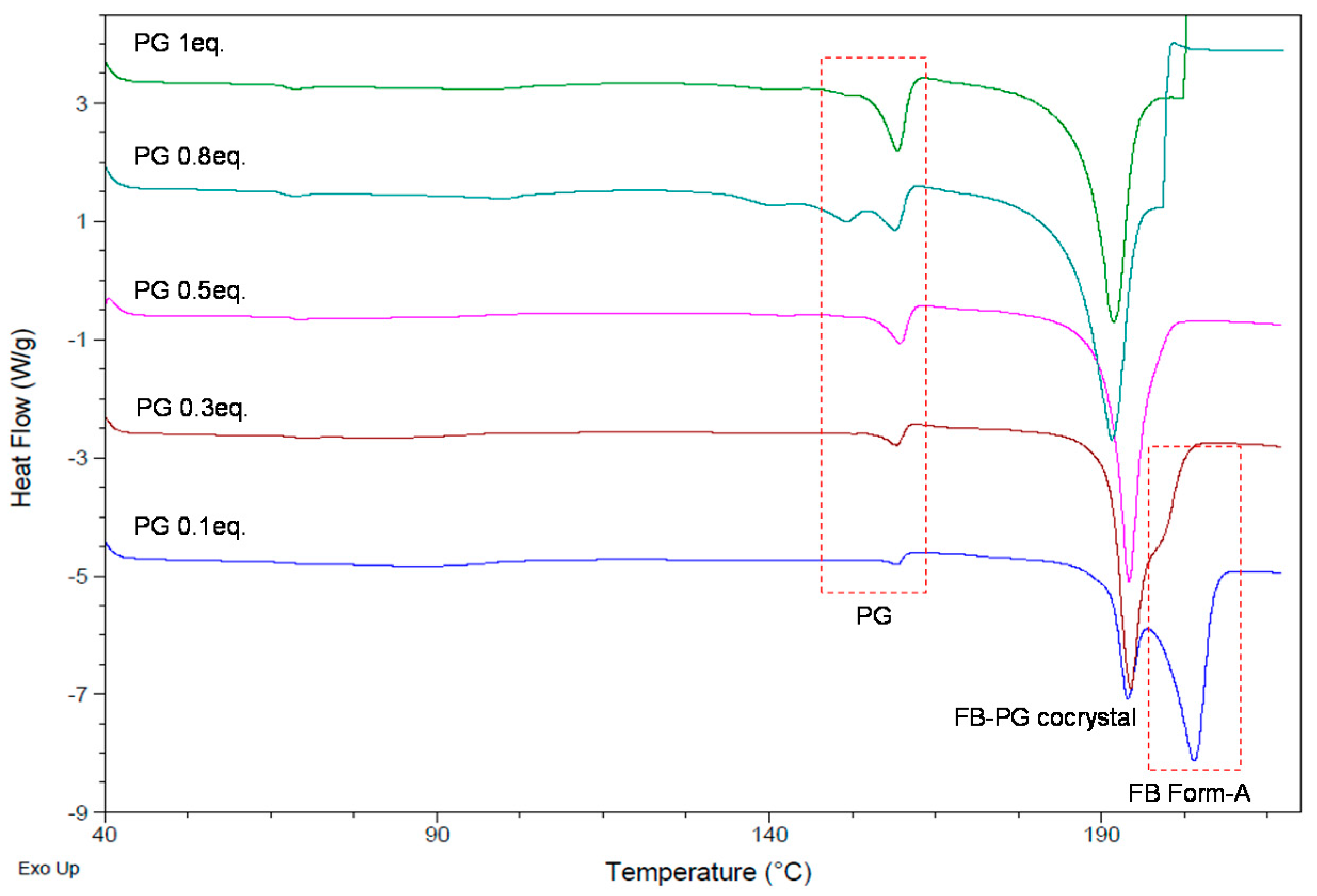
2.1. Cocrystal Screening of Febuxostat
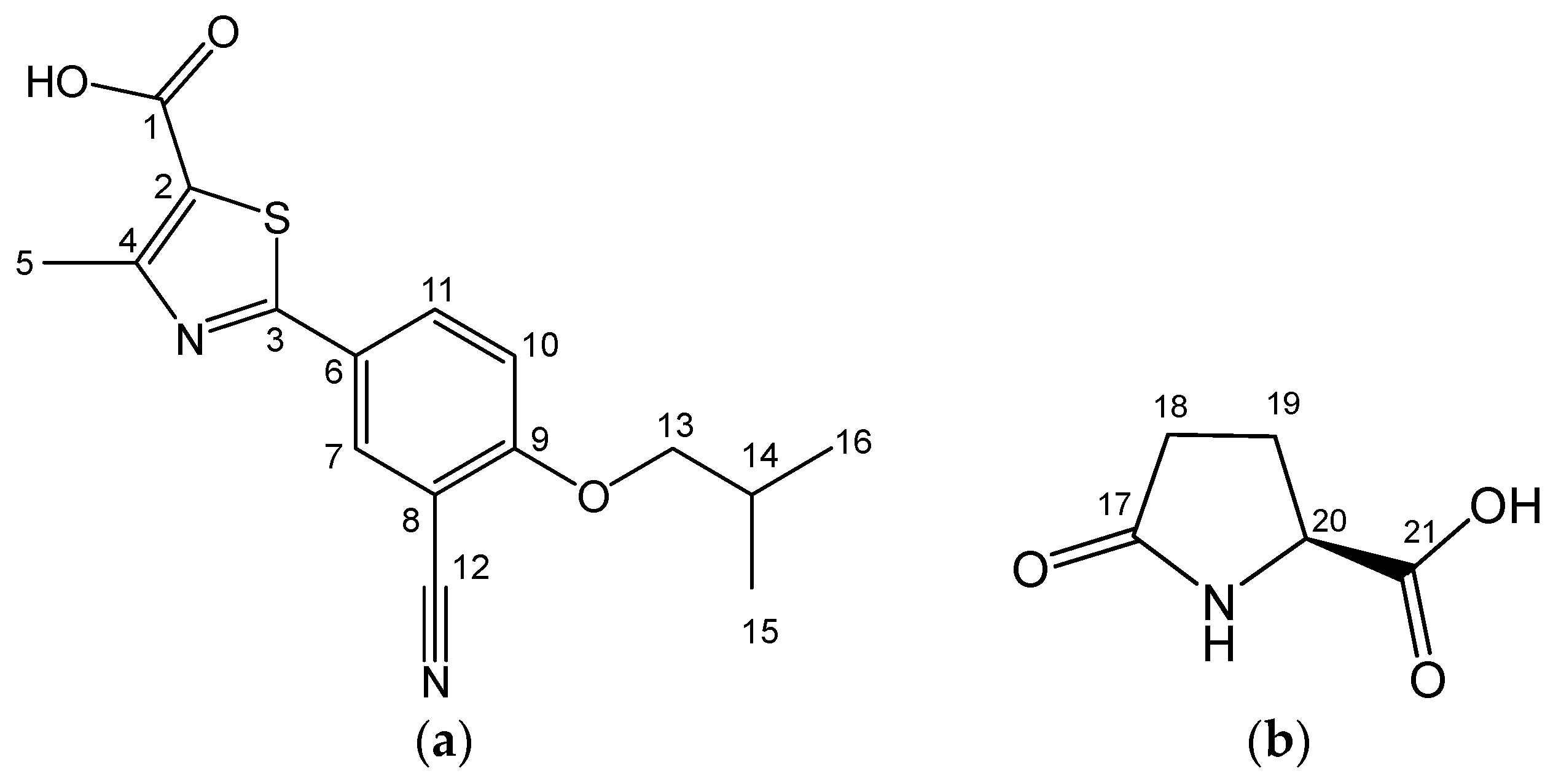
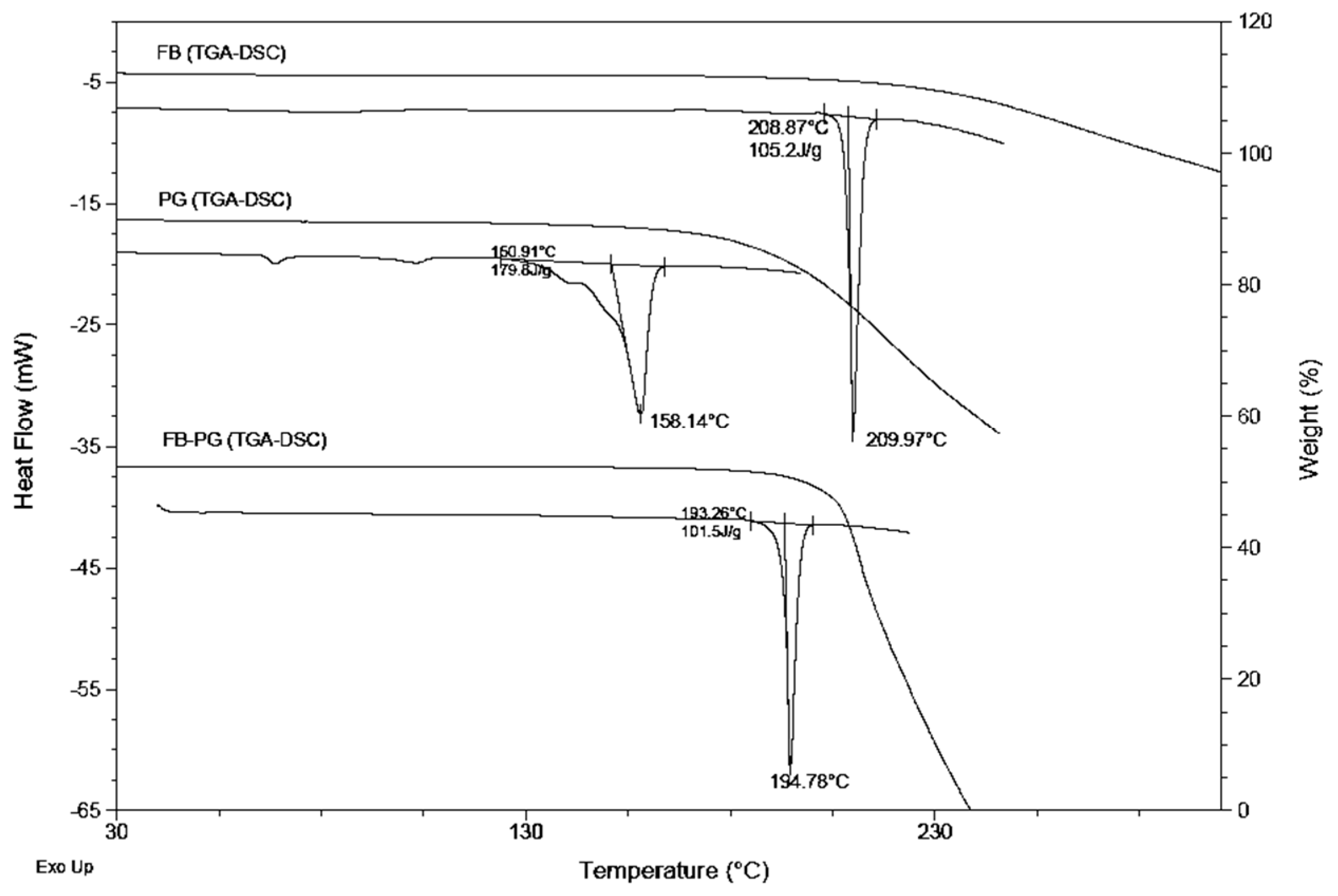
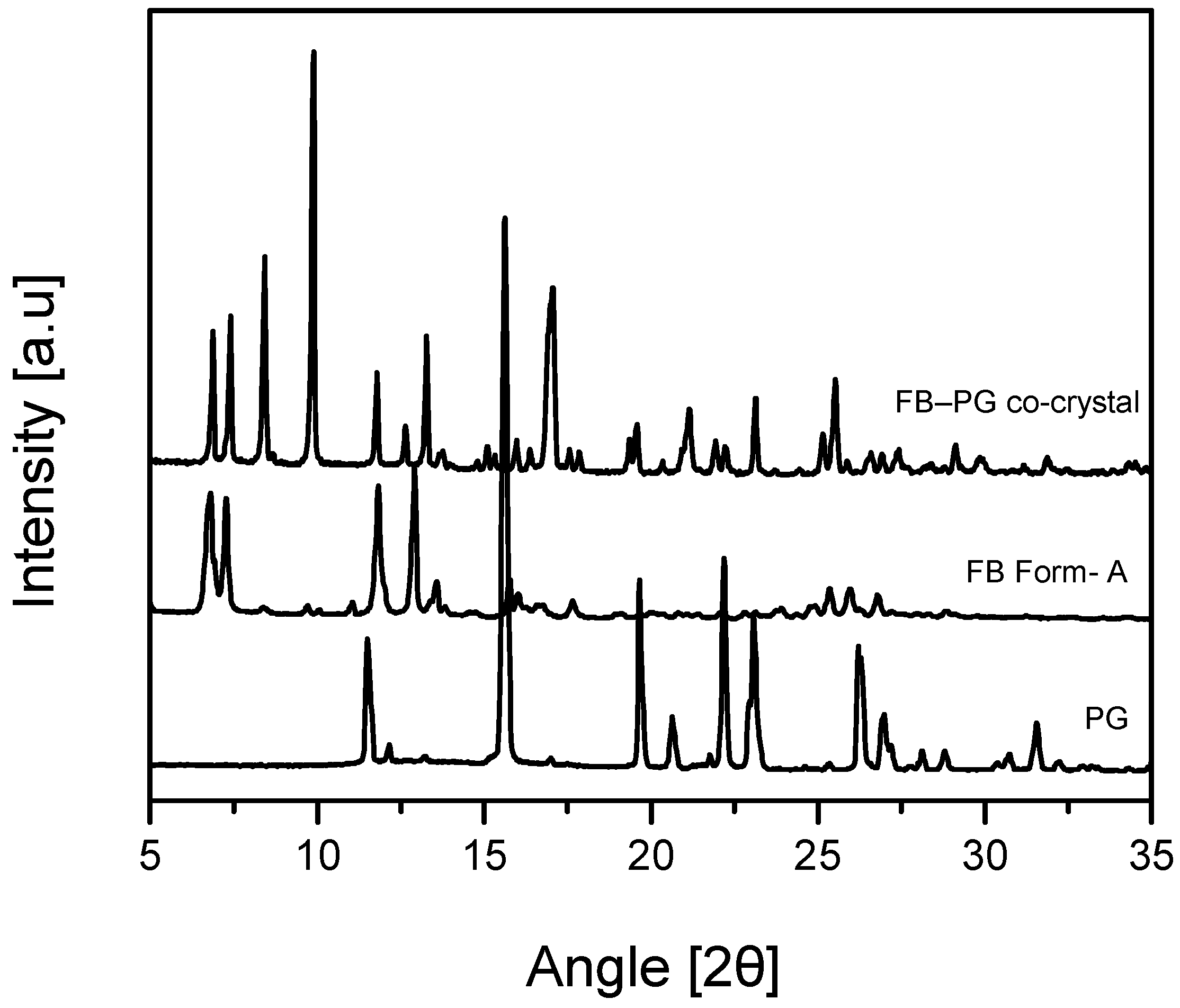
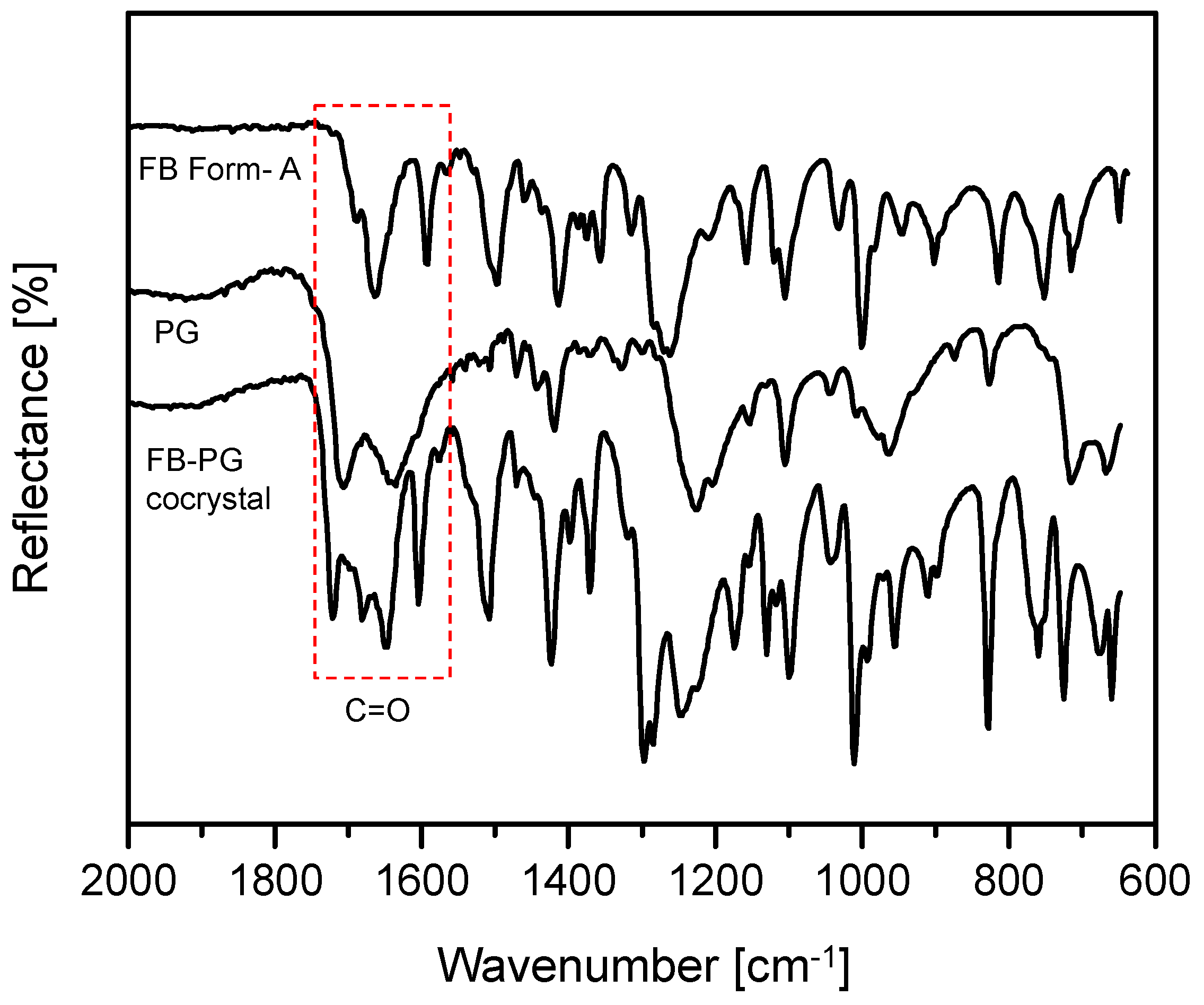
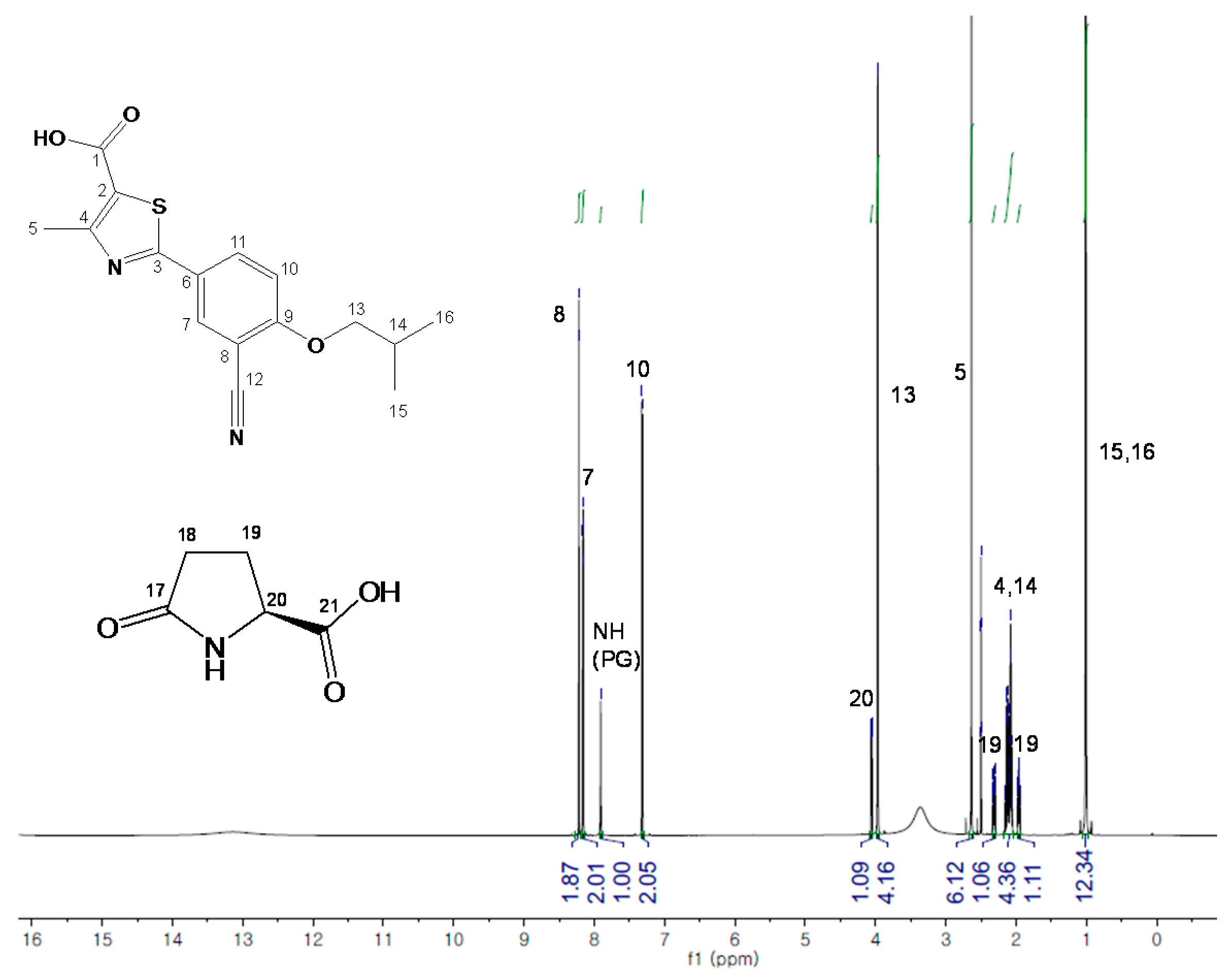
2.2. FB-PG Cocrystal Characterization
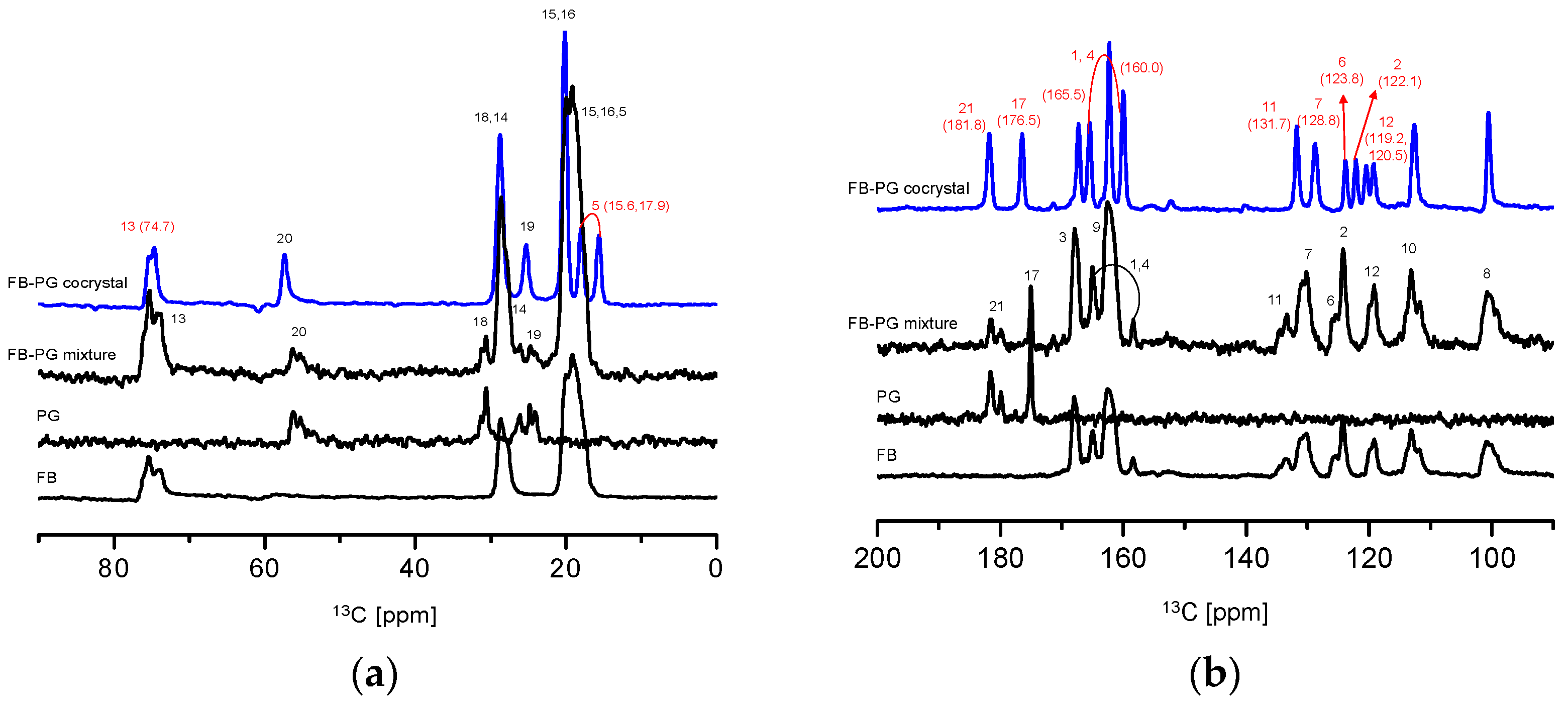
2.3. Prediction of FB-PG Cocrystal’s Intermolecular Interactions Using Solid-State CP/MAS 13C-NMR
2.4. Solubility of FB-PG Cocrystal
3. Materials and Methods
3.1. Materials
3.2. Production of FB-PG Cocrystal by Using Liquid-Assisted Grinding Technique
3.3. Solid-State Nuclear Magnetic Resonance Spectroscopy (Solid-State CP/MAS 13C-NMR)
3.4. Solution-State Nuclear Magnetic Resonance Spectroscopy (Solution-State NMR)
3.5. Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR)
3.6. Powder X-ray Diffraction (PXRD)
3.7. Differential Scanning Calorimetry (DSC)
3.8. Solubility Test
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hilfiker, R. Polymorphism in the Pharmaceutical Industry; Wiley-Vch: Weinheim, Germany, 2006; pp. 1–15. [Google Scholar]
- Chen, J.; Sarma, B.; Myerson, A.S. Pharmaceutical crystallization. Cryst. Growth Des. 2011, 11, 887–895. [Google Scholar] [CrossRef]
- Sparma, B.; Chen, J.; His, H.Y.; Myerson, A.S. Solid form of pharmaceuticals: Polymorphs, salt and cocrystals. Korean J. Chem. Eng. 2011, 28, 315–322. [Google Scholar] [CrossRef]
- Babu, N.J.; Nangia, A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Reddy, L.S.; Babu, N.J.; Nangia, A. Carboxamide-pyridine N-oxide heterosynthon for crystal engineering and pharmaceutical cocrystals. Chem. Commun. 2006, 13, 1369–1371. [Google Scholar] [CrossRef] [PubMed]
- Bhogala, B.R.; Basavoju, S.; Nangia, A. Three-component carboxylic acid-bipyridine lattice inclusion host. Supramolecular synthesis of ternary cocrystals. Cryst. Growth Des. 2005, 5, 1683–1686. [Google Scholar] [CrossRef]
- Reddy, L.S.; Basavoju, S.; Vangala, V.R.; Nangia, A. Hydrogen bonding in crystal structures of N,N′-Bis (3-pyridyl) urea. Why is the NH tape synthon absent in diaryl ureas with electron-withdrawing groups? Cryst. Growth Des. 2006, 6, 161–173. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Hussain, I.; Desper, J. 2-Acetaminopyridine: A highly effective cocrystallizing agent. Cryst. Growth Des. 2006, 6, 474–480. [Google Scholar] [CrossRef]
- Chadha, R.; Bhalla, Y.; Vashisht, M.K.; Chadha, K. Recrystallization in Materials Processing; Intech: Vienna, Austria, 2015; pp. 173–174. [Google Scholar]
- Harris, R.K. Application of solid-state NMR to pharmaceutical polymorphism and related matters. J. Pharm. Pharmacol. 2007, 59, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Vogt, F.G.; Clawson, J.S.; Strohmeier, M.; Edwards, A.J.; Pham, T.N.; Watsom, S.A. Solid-State NMR analysis of organic cocrystals and complexes. Cryst. Growth Des. 2009, 9, 921–937. [Google Scholar] [CrossRef]
- Mandala, V.S.; Loewus, S.J.; Mehta, M.A. Monitoring cocrystal formation via in situ solid-state NMR. J. Phys. Chem. Lett. 2014, 5, 3340–3344. [Google Scholar] [CrossRef] [PubMed]
- Sokal, A.; Pindelska, E.; Szeleszczuk, L.; Kolodziejski, W. Pharmaceutical properties of two ethenzamide-gentisic acid cocrystal polymorphs: Drug release profiles, spectroscopic studies and theoretical calculations. Int. J. Pharm. 2017, 522, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Watanabe, K.; Hiramatsu, T.; Kitamura, M. Polymorphs of 2-(3-cyano-4-isovutyloxyphenyl)-4-methyl-5-thiazolecarboxylic Acid and Method of Producing the Same. U.S. Patent 6225474 B1, 1 May 2001. [Google Scholar]
- Iwai, M.; Nakamura, K.; Dohi, M.; Mochizuki, H.; Mochizuki, S. Solid Preparation Containing Single Crystal Form. U.S. Patent 7361676 B2, 22 April 2008. [Google Scholar]
- Jiang, Q.Y.; Qian, J.J.; Gu, J.M.; Tang, G.P.; Hu, X.R. Febuxostat methanol solvate. Acta Cryst. 2011, E67, 1232. [Google Scholar] [CrossRef] [PubMed]
- Maddileti, D.; Jayabun, S.K.; Nangia, A. soluble cocrystals of the xanthine oxidase inhibitor febuxostat. Cryst. Growth Des. 2013, 13, 3188–3196. [Google Scholar] [CrossRef]
- Wu, M.; Hu, X.R.; Gu, J.M.; Tang, G.P. Crystal structure of febuxostat-acetic acid (1/1). Acta Cryst. 2015, E71, 295–296. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.H.; Wermuth, C.G. Handbook of Pharmaceutical Salt Properties, Selection, and Use; Wiley-Vch: Weinheim, Germany, 2002; pp. 272–320. [Google Scholar]
- Vallu, V.R.; Desai, K.G.; Patil, S.D.; Agarwal, R.; Padi, P.R.; Ghanta, M.R. Synthesis and characterization of some analogues of Febuxostat—An anti-hyperuricemia drug. ACAIJ 2014, 14, 339–350. [Google Scholar]
- Vallu, V.R.; Desai, K.G.; Patil, S.D.; Agarwal, R.; Padi, P.R.; Ghanta, M.R. Synthesis and characterization of process-related impurities of an anti-hyperuricemia drug-Febuxostat. Der Pharma Chem. 2014, 6, 300–311. [Google Scholar]
- Yoshifuji, S.; Tanaka, K.I.; Kawal, T.; Nitta, Y. A novel of l-pyroglutamic acid derivatives from l-proline: utility of N-protecting groups for ruthenium tetroxide oxidation of cyclic α-amino acids. Chem. Pharm. Bull. 1986, 34, 3873–3878. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coformer | pKa [19] |
|---|---|
| l-Glutamic acid (1 eq.) | 2.19 |
| l-Lysine (1 eq.) | 2.16 |
| l-Aspartic acid (1 eq.) | 1.88 |
| l-Pyroglutamic Acid (PG) (1 eq.) | 3.32 |
| l-Arginine (1 eq.) | 2.18 |
| FB form-A [μmol/mL] | FB-PG Cocrystal [μmol/mL] | |
|---|---|---|
| water | 0.041 | 0.161 |
| pH 1.2 | 1.337 | 2.605 |
| pH 4.0 | 0.129 | 0.670 |
| pH 6.8 | 0.041 | 0.170 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, J.-H.; Lim, C.; Ryu, H.C.; Kim, J.S.; Kim, H.M.; Kiyonga, A.N.; Park, M.; Suh, Y.-G.; Park, G.H.; Jung, K. Structural Characterization of Febuxostat/l-Pyroglutamic Acid Cocrystal Using Solid-State 13C-NMR and Investigational Study of Its Water Solubility. Crystals 2017, 7, 365. https://doi.org/10.3390/cryst7120365
An J-H, Lim C, Ryu HC, Kim JS, Kim HM, Kiyonga AN, Park M, Suh Y-G, Park GH, Jung K. Structural Characterization of Febuxostat/l-Pyroglutamic Acid Cocrystal Using Solid-State 13C-NMR and Investigational Study of Its Water Solubility. Crystals. 2017; 7(12):365. https://doi.org/10.3390/cryst7120365
Chicago/Turabian StyleAn, Ji-Hun, Changjin Lim, Hyung Chul Ryu, Jae Sun Kim, Hyuk Min Kim, Alice Nguvoko Kiyonga, Minho Park, Young-Ger Suh, Gyu Hwan Park, and Kiwon Jung. 2017. "Structural Characterization of Febuxostat/l-Pyroglutamic Acid Cocrystal Using Solid-State 13C-NMR and Investigational Study of Its Water Solubility" Crystals 7, no. 12: 365. https://doi.org/10.3390/cryst7120365
APA StyleAn, J. -H., Lim, C., Ryu, H. C., Kim, J. S., Kim, H. M., Kiyonga, A. N., Park, M., Suh, Y. -G., Park, G. H., & Jung, K. (2017). Structural Characterization of Febuxostat/l-Pyroglutamic Acid Cocrystal Using Solid-State 13C-NMR and Investigational Study of Its Water Solubility. Crystals, 7(12), 365. https://doi.org/10.3390/cryst7120365