
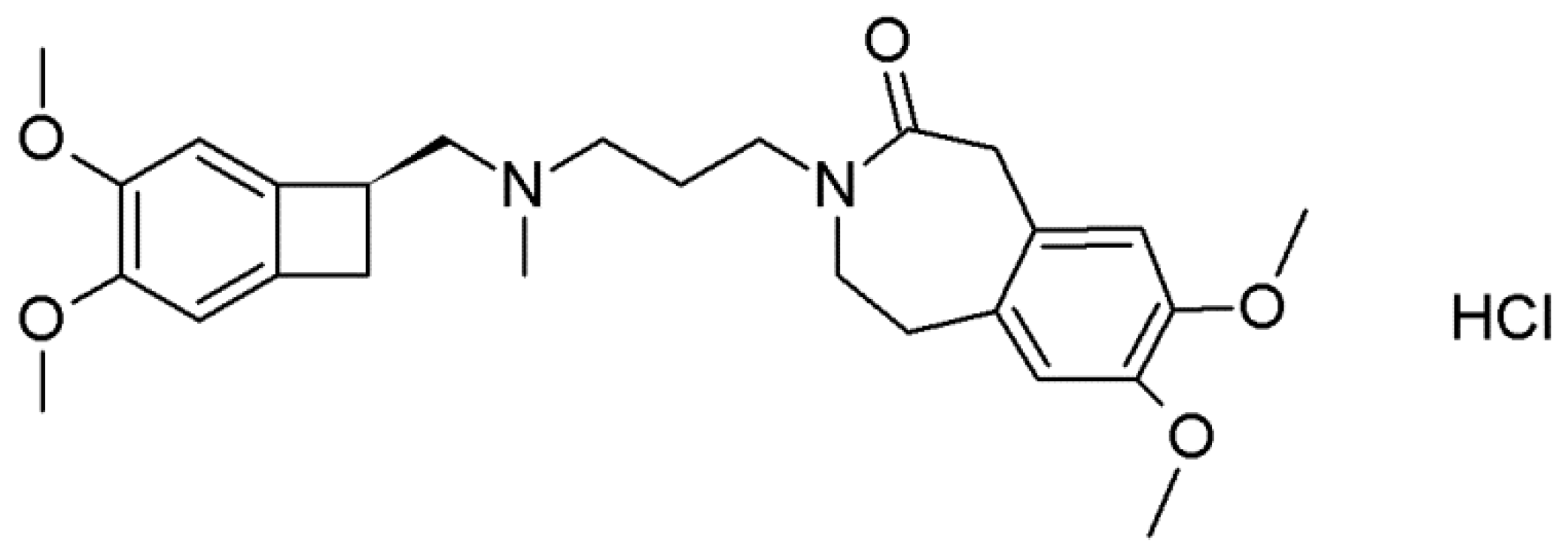
Ivabradine Hydrochloride (S)-Mandelic Acid Co-Crystal: In Situ Preparation during Formulation

Abstract
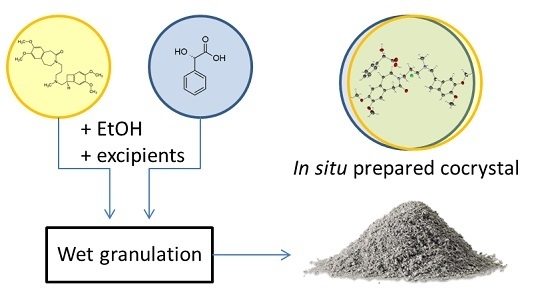
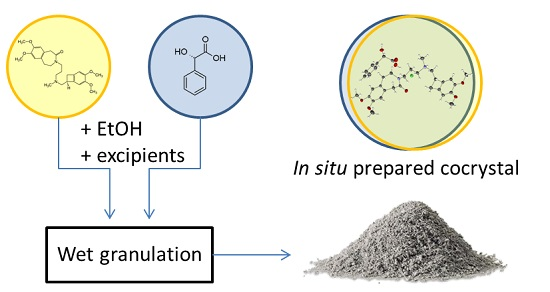
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Analytical Methods
2.2.1. Powder X-ray Diffraction (XRPD)
2.2.2. Infrared (IR) Spectroscopy
2.2.3. Raman Spectroscopy
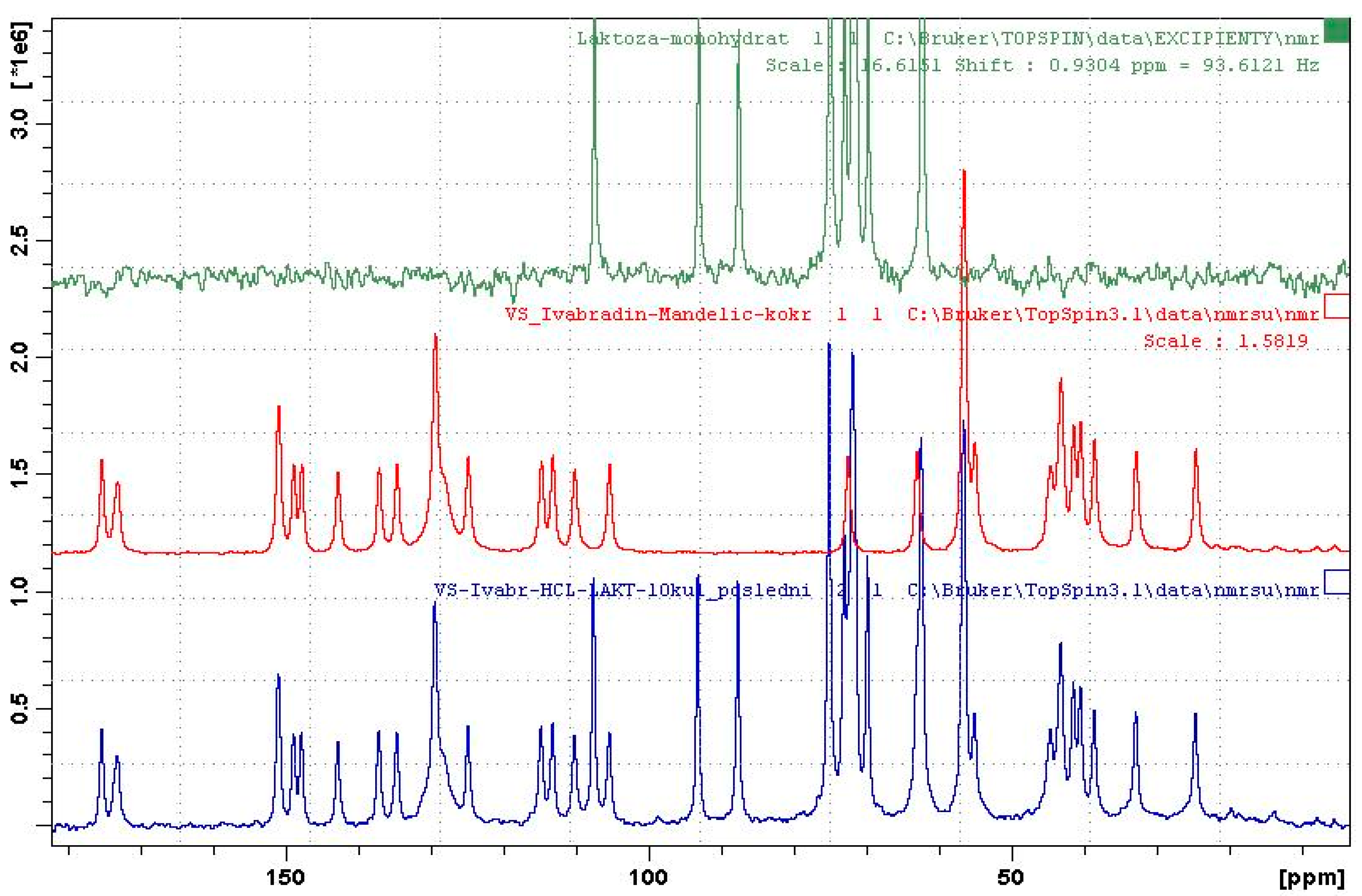
2.2.4. Solution Nuclear Magnetic Resonance (Solution NMR)
2.2.5. Solid-State Nuclear Magnetic Resonance (ssNMR)
2.2.6. Differential Scanning Calorimetry (DSC)
2.2.7. Dynamic Vapor Sorption (DVS)
2.2.8. Karl Fischer Coulometric Titration
2.2.9. Single Crystal X-ray Diffraction (SXRD)
2.2.10. Ultra Performance Liquid Chromatography (UPLC)
2.3. Sample Preparation
2.3.1. Co-Crystal Screening
2.3.2. Co-Crystal Reproduction
2.3.3. Maturation Experiments
2.3.4. Long-Term Physical Stability Studies of IClSM
2.3.5. Physical and Chemical Stability: Stress Studies
2.3.6. Pre-Formulation of Ivabradine Hydrochloride (S)-Mandelic Acid Co-Crystal (IClSM)
2.4. Formulation
Characterization of Mixtures A–J
3. Results and Discussion
3.1. Co-Crystal Screening and Solid-State Characterization of Hits
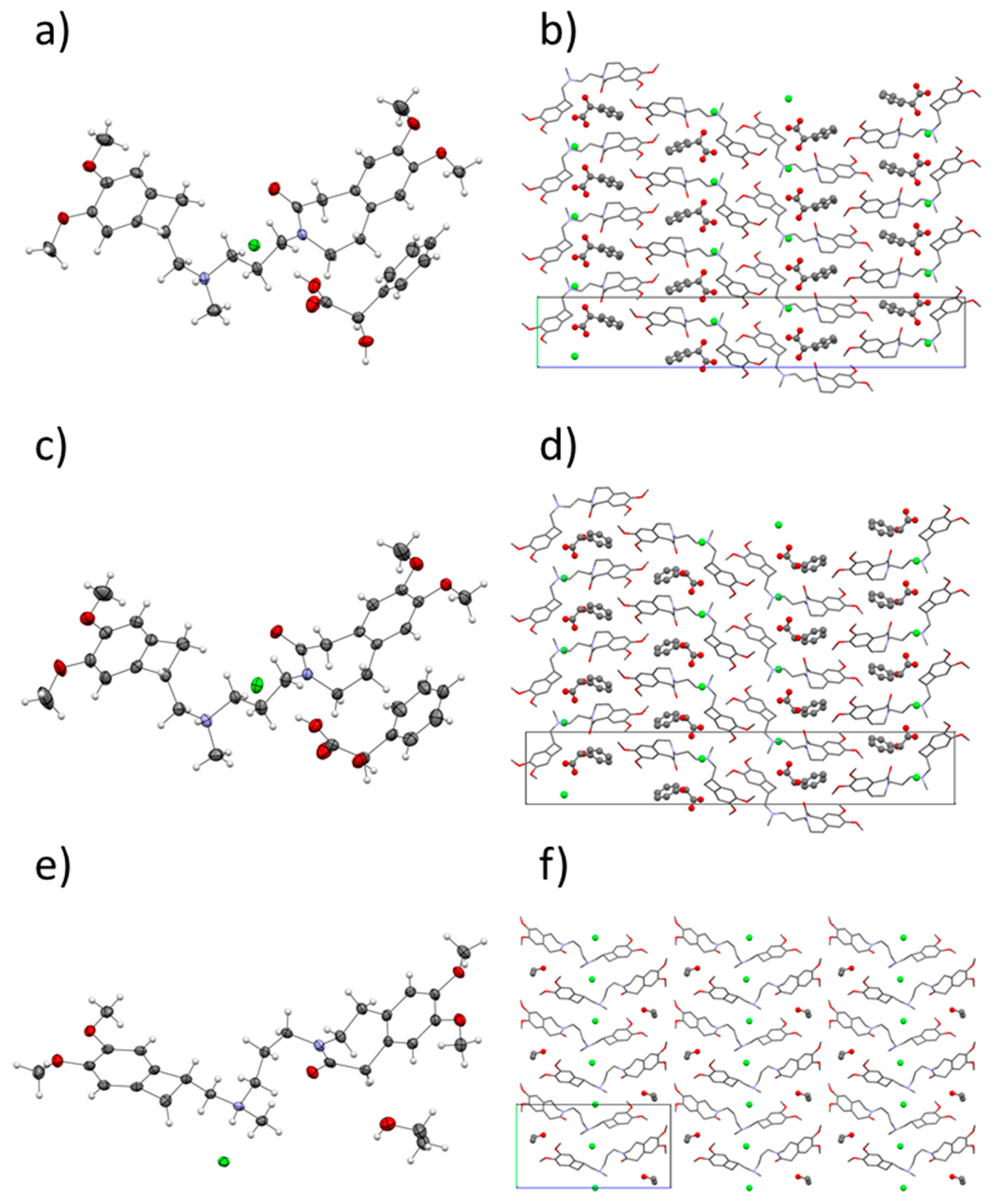
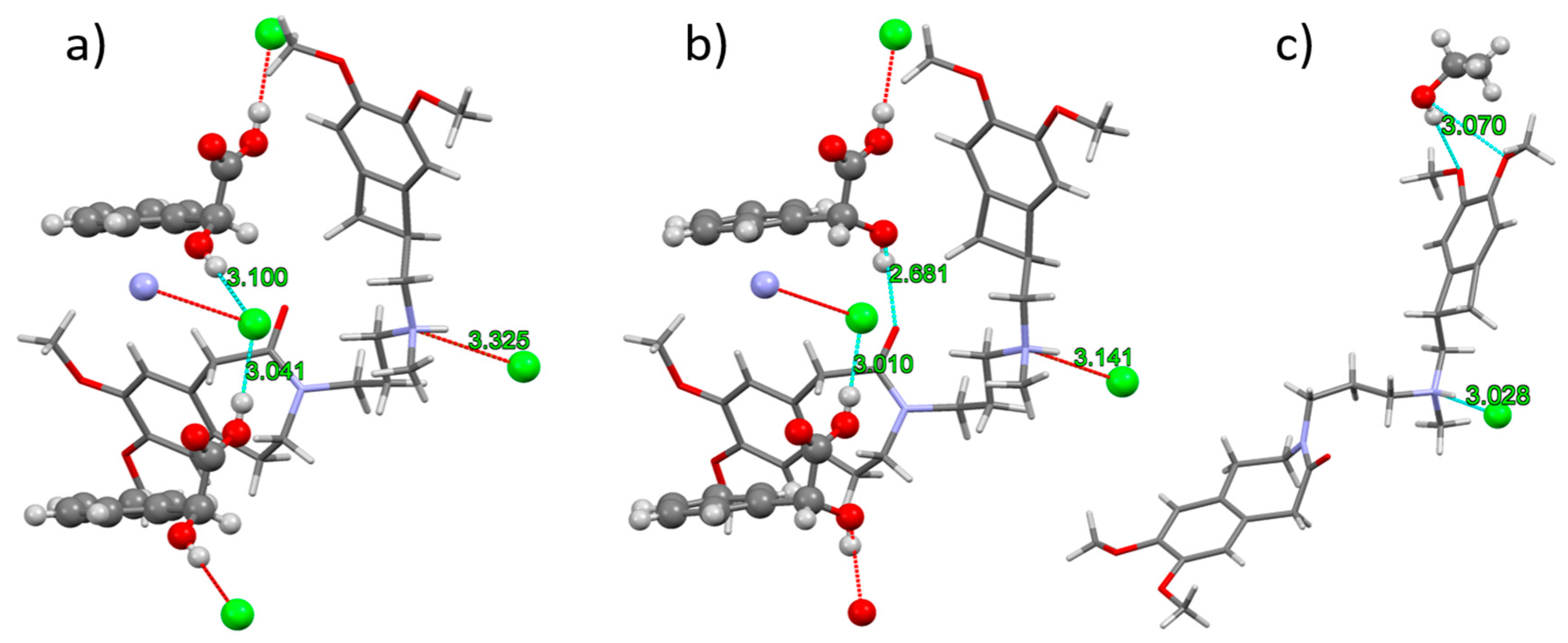
3.2. Crystal Structures and Their Comparison
3.3. Co-Crystal Selection and Comparison with Other Forms of IVA HCl
3.4. In Situ Formation of the Co-Crystal during Wet Granulation
3.5. Comparison of Pre-Prepared and in Situ Formed Co-Crystal in Formulation and Selection of Final Excipients Composition
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wouters, J.; Quere, L. Pharmaceutical Salts and Co-Crystals 2012; Royal Society of Chemistry: Cambridge, UK, 2012; p. 391. [Google Scholar]
- Friščic, T.; Jones, W. Recent Advances in Understanding the Mechanism of Cocrystal Formation via Grinding. Cryst. Growth Des. 2009, 9, 1621–1637. [Google Scholar] [CrossRef]
- Etter, M.C.; Reutzel, S.M.; Choo, C.G. Self-organization of adenine and thymine in the solid state. J. Am. Chem. Soc. 1993, 115, 4411–4412. [Google Scholar] [CrossRef]
- Trask, A.V.; Motherwell, W.D.S.; Jones, W. Solvent-drop grinding: Green polymorph control of cocrystallization. Chem. Commun. 2004, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.J.; Horton, P.N.; Hursthouse, M.B.; Storey, R.; Jones, W.; Friščić, T.; Seaton, C.C.; Clegg, W.; Harrington, R.W.; Coles, S.J. Applying Hot-Stage Microscopy to Co-Crystal Screening: A Study of Nicotinamide with Seven Active Pharmaceutical Ingredients. Cryst. Growth Des. 2008, 8, 1697–1712. [Google Scholar] [CrossRef]
- Lu, E.; Rodriguez-Hornedo, N.; Suryanarayanan, R. A rapid thermal method for cocrystal screening. CrystEngComm 2008, 10, 665–668. [Google Scholar] [CrossRef]
- Childs, S.L.; Rodriguez-Hornedo, N.; Reddy, L.S.; Jayasankar, A.; Maheshwari, C.; McCausland, L.; Shipplett, R.; Stahly, B.C. Screening strategies based on solubility and solution composition generate pharmaceutically acceptable cocrystals of carbamazepine. CrystEngComm 2008, 10, 856–864. [Google Scholar] [CrossRef]
- Duggirala, N.K.; Perry, M.L.; Almarsson, O.; Zaworotko, M.J. Hydrogen Bond Hierarchy: Persistent Phenol··· Chloride Hydrogen Bonds in the Presence of Carboxylic Acid Moieties. Cryst. Growth Des. 2015, 15, 4341–4354. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Maini, L.; Prosperi, S.; Gobetto, R.; Chierotti, M.R. From unexpected reactions to a new family of ionic co-crystals: The case of barbituric acid with alkali bromides and caesium iodide. Chem. Commun. (Camb.) 2010, 46, 7715–7717. [Google Scholar] [CrossRef] [PubMed]
- Sladkova, V.; Skalicka, T.; Skorepova, E.; Cejka, J.; Eigner, V.; Kratochvil, B. Systematic solvate screening of trospium chloride: Discovering hydrates of a long-established pharmaceutical. CrystEngComm 2015, 17, 4712–4721. [Google Scholar] [CrossRef]
- Tieger, E.; Kiss, V.; Pokol, G.; Finta, Z.; Dušek, M.; Rohlíček, J.; Skořepová, E.; Brázda, P. Studies on the crystal structure and arrangement of water in sitagliptin l-tartrate hydrates. CrystEngComm 2016, 18, 3819–3831. [Google Scholar] [CrossRef]
- Childs, S.L. Crystal engineering approach to forming cocrystals of amine hydrochlorides with organic acids. Molecular complexes of fluoxetine hydrochloride with benzoic, succinic, and fumaric acids. J. Am. Chem. Soc. 2004, 126, 13335–13342. [Google Scholar] [CrossRef] [PubMed]
- Sládková, V.; Cibulková, J.; Eigner, V.; Šturc, A.; Kratochvíl, B.; Rohlíček, J. Application and Comparison of Cocrystallization Techniques on Trospium Chloride Cocrystals. Cryst. Growth Des. 2014, 14, 2931–2936. [Google Scholar] [CrossRef]
- Skorepova, E.; Husak, M.; Cejka, J.; Zamostny, P.; Kratochvil, B. Increasing dissolution of trospium chloride by co-crystallization with urea. J. Cryst. Growth 2014, 399, 19–26. [Google Scholar] [CrossRef]
- Brittain, H.G. Pharmaceutical cocrystals: The coming wave of new drug substances. J. Pharm. Sci. 2013, 102, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Pop, M.; Sieger, P.; Cains, P.W. Tiotropium fumarate: An interesting pharmaceutical co-crystal. J. Pharm. Sci. 2009, 98, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and fictions about polymorphism. Chem. Soc. Rev. 2015, 44, 8619–8635. [Google Scholar] [CrossRef] [PubMed]
- Prohens, R.; Barbas, R.; Portell, A.; Font-Bardia, M.; Alcobé, X.; Puigjaner, C. Polymorphism of Cocrystals: The Promiscuous Behavior of Agomelatine. Cryst. Growth Des. 2016, 16, 1063–1070. [Google Scholar] [CrossRef]
- Peglion, J.L.; Vian, J.; Vilaine, J.P.; Villeneuve, N.; Janiak, P.; Bidouard, J.P. Preparation of N-[(Benzocyclobutylalkyl)amino]alkyl]tetrahydro-3-benzazepin-2-ones and Analogs as Cardiovascular Agents; Adir et Compagnie Patents: Courbevoie, France, 1993; p. 24. [Google Scholar]
- Horvath, S.; Auguste, M.-N.; Damien, G. Process for the Preparation of the Polymorphic γ-Crystalline Form of Ivabradine Hydrochloride and Pharmaceutical Compositions Containing It; Les Laboratoires Servier: Suresnes, France, 2006; p. 4. [Google Scholar]
- Lerestif, J.-M.; Lecouve, J.-P.; Souvie, J.-C.; Brigot, D.; Horvath, S.; Auguste, M.-N.; Damien, G. Process for the Preparation of Ivabradine via Hydrogenation of Oxobenzazepinepropanal Acetal Derivative and Coupling with Dimethoxycyclobutabenzenemethanamine; Les Laboratoires Servier: Suresnes, France, 2005; p. 6. [Google Scholar]
- Horvath, S.; Auguste, M.-N.; Damien, G. Preparation of β-Crystalline Form of Ivabradine Hydrochloride for Pharmaceuticals; Les Laboratoires Servier: Suresnes, France, 2006; p. 9. [Google Scholar]
- Horvath, S.; Auguste, M.-N.; Damien, G. Preparation of β-d Crystalline Form of Ivabradine Hydrochloride for Pharmaceuticals; Les Laboratoires Servier: Suresnes, France, 2006; p. 8. [Google Scholar]
- Horvath, S.; Auguste, M.-N.; Damien, G. Preparation of γ-d Crystalline Form of Ivabradine Hydrochloride for Pharmaceutical Compositions; Les Laboratoires Servier: Suresnes, France, 2006; p. 9. [Google Scholar]
- Horvath, S.; Auguste, M.-N.; Damien, G. Delta Crystalline Form of Ivabradine Hydrochloride; Les Laboratoires Servier: Suresnes, France, 2007; p. 4. [Google Scholar]
- Barreca, G.; Gatti, M.M.; Ventimiglia, G. New Polymorph of Ivabradine Hydrochloride and Method for Its Preparation; Chemo Research, S.L.: Madrid, Spain, 2014; p. 25. [Google Scholar]
- Dwivedi, S.D.; Prasad, A.; Patel, M.S.; Sharma, P.R. Preparation of a Polymorphic Form of Ivabradine Hydrochloride; Chemical Indexing Equivalent to 160:166199 (IN); Cadila Healthcare Limited: Ahmedabad, India, 2012; p. 29. [Google Scholar]
- Dwivedi, S.D.; Kumar, R.; Patel, S.T.; Shah, A.P.C. Process for Preparation of Ivabradine Hydrochloride; Cadila Healthcare Limited: Ahmedabad, India, 2008; p. 33. [Google Scholar]
- Dwivedi, S.D.; Prasad, A.; Sharma, M.H.; Sharma, P.R.; Parihar, J.A. Polymorphic Forms of Ivabradine Hydrochloride; Chemical Indexing Equivalent to 159:172881 (IN); Cadila Healthcare Limited: Ahmedabad, India, 2013; p. 35. [Google Scholar]
- Kotar-Jordan, B.; Gojak, U.; Smrkolj, M. Novel Forms of Ivabradine Hydrochloride; Krka: Novo Mesto, Slovenia, 2011; p. 36. [Google Scholar]
- Chen, Y.; Hong, C.; Liu, D.; Zhu, W. Ivabradine Hydrochloride New Crystal Form C, and Its Preparation Method; Jiangsu Yutian Bio-Pharmaceutical Technology Co., Ltd.: Lianyungang, China, 2013; p. 9. [Google Scholar]
- Zhang, X.; Cui, D.; Zhang, T. S Crystal Form of Ivabradine Hydrochloride, and Prepn. Method and Pharmaceutical Compn; Beijing Lunarsun Pharmaceutical Co., Ltd.: Beijing, China, 2015; p. 23. [Google Scholar]
- Prohens Lopez, R.; Puigjaner Vallet, C.; Barbas Canero, R.; Del Rio Pericacho, J.L.; Marti, J. Preparation of Ivabradine Hydrochloride Form IV; Urquima S.A.: Barcelona, Spain, 2013; p. 30. [Google Scholar]
- Wieser, J.; Griesser, U.; Enders, M.; Kahlenberg, V. Preparation of Acetone Solvate of Ivabradine Hydrochloride; Sandoz AG: Rotkreuz ZG, Switzerland, 2013; p. 44. [Google Scholar]
- Sun, P.; Chen, Y.; Yu, G. Ivabradine Sulfate, Its Type I Crystal and Preparation Method Thereof; Jiangsu Hengrui Medicine Co., Ltd.: Lianyungang, China, 2010; p. 8. [Google Scholar]
- Singh, S.P.; Singh, G.; Wadhwa, L. Process for the Preparation of Ivabradine Hydrochloride and Related Polymorphs; IND-Swift Laboratories Limited: Chandigarh, India, 2008; p. 22. [Google Scholar]
- Singh, G.; Singh, S.P.; Wadhwa, L. Acid Addition Salts of Ivabradine and Preparation Thereof; IND-Swift Laboratories Limited: Chandigarh, India, 2011; p. 24. [Google Scholar]
- Gidwani, R.M.; Kolhatkar, M.V.; Meergans, D.; Stefan, R.; Geier, J. Ivabradine-Containing Pharmaceutical Composition with Modified Release; Ratiopharm GmbH: Ulm, Germany, 2011; p. 28. [Google Scholar]
- Smrkolj, M.; Gojak, U.; Kotar-Jordan, B. Pharmaceutical Compositions Comprising Ivabradine Hydrobromide; Krka: Tovarna Zdravil, d.d., Slovenia, 2009; p. 34. [Google Scholar]
- Alhalaweh, A.; Velaga, S.P. Formation of Cocrystals from Stoichiometric Solutions of Incongruently Saturating Systems by Spray Drying. Cryst. Growth Des. 2010, 10, 3302–3305. [Google Scholar] [CrossRef]
- Daurio, D.; Medina, C.; Saw, R.; Nagapudi, K.; Alvarez-Núñez, F. Application of Twin Screw Extrusion in the Manufacture of Cocrystals, Part I: Four Case Studies. Pharmaceutics 2011, 3, 582–600. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Motherwell, W.D.S. Applications of the Cambridge Structural Database in organic chemistry and crystal chemistry. Acta Crystallogr. Sect. B 2002, 58, 407–422. [Google Scholar] [CrossRef]
- Duval, D.; Hennig, P.; Bouchet, J.P.; Vian, J.; Peglion, J.L.; Volland, J.P.; Platzer, N.; Guilhem, J. Stereochemical Study of a Bradicardisant Benzazepine-Type Drug. X-ray Structure of the Chloride Salt and High-Field NMR Study of the Stereochemistry in Solution. Magn. Reson. Chem. 1997, 35, 175–183. [Google Scholar] [CrossRef]
- Hu, X.; Gu, J.; Jin, Z.; Huang, Y.; Huang, J. Stable Crystal Form II of Ivabradine Hydrochloride and Preparation Method Thereof; Zhejiang Jingxin Pharmaceutical Co., Ltd.: Shaoxing, China, 2013; p. 40. [Google Scholar]
- Masciocchi, N.; Aulisio, A.; Bertolini, G.; Sada, M.; Garis, F.; Malpezzi, L. Disclosing the extensive crystal chemistry of Ivabradine hydrochloride, in its pure and solvated phases. Powder Diffr. 2013, 28, 200–206. [Google Scholar] [CrossRef]
- Council of Europe. European Pharmacopoeia, 7th ed.; Council of Europe: Strasbourg, France, 2012. [Google Scholar]
- Skorepova, E.; Husak, M.; Ridvan, L.; Tkadlecova, M.; Havlicek, J.; Dusek, M. Iodine salts of a pharmaceutical compound agomelatine: Effect of symmetric H-bond on amide protonation. CrystEngComm 2016, 18, 4518–4529. [Google Scholar] [CrossRef]
- Lubach, J.W.; Xu, D.; Segmuller, B.E.; Munson, E.J. Investigation of the effects of pharmaceutical processing upon solid-state NMR relaxation times and implications to solid-state formulation stability. J. Pharm. Sci. 2007, 96, 777–787. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | IVA HCl (mg) | (S)-Mandelic Acid (mg) | Lactose Monohydrate (mg) | Solvent | |
|---|---|---|---|---|---|
| Type | Amount (µL) | ||||
| Liquid-assisted grinding (LAG) | 100 | 30 | - | EtOH | 50 |
| 100 | 30 | - | WA | 40 | |
| 200 | 60 | - | WA | 20 | |
| Mixing in a vial by microspatula | 10 | 3 | 130 | EtOH | 30 |
| 10 | 3 | 130 | EtOH | 60 | |
| 10 | 3 | 130 | EtOH | 100 | |
| 10 | 3 | 130 | WA | 30 | |
| 10 | 3 | 130 | WA | 70 | |
| 100 | 30 | 1300 | EtOH | 50 | |
| 100 | 30 | 1300 | EtOH | 100 | |
| 100 | 30 | 1300 | EtOH | 150 | |
| 100 | 30 | 1300 | EtOH | 200 | |
| High-shear granulation * | 300 | 90 | 4000 | EtOH | 1200 |
| 300 | 90 | 4000 | EtOH | 1800 | |
| 300 | 90 | 4000 | EtOH | 2100 | |
| Mixing in a beaker | 750 | 225 | 10,000 | EtOH | 560 |
| Ingredient (mg) | A | B | C |
|---|---|---|---|
| IClSM | 10.5 | 10.5 | 10.5 |
| Klucel EF | 2.7 | 2.7 | 2.7 |
| Lactose monohydrate | 115.0 | - | 111.9 |
| Mannitol | - | 115 | - |
| Primojel type A | 5.4 | 5.4 | 5.4 |
| Meglumine | - | - | 3.125 |
| Magnesium stearate | 1.35 | 1.35 | 1.35 |
| Tablet weight (mg) | 135 | 135 | 135 |
| Ingredient (mg) | D | E | F | G | H | I | J |
|---|---|---|---|---|---|---|---|
| Ivabradine HCl | 8.085 | 8.085 | 8.085 | 8.085 | 8.085 | 8.085 | 8.085 |
| (S)-mandelic acid | 2.436 | 2.436 | 2.436 | 2.436 | 2.436 | 2.436 | 2.436 |
| Klucel EF | 2.7 | 2.7 | 2.7 | 2.7 | 2.7 | 2.7 | 2.7 |
| Lactose monohydrate | 115.0 | - | 112.3 | 114.25 | 111.9 | 114.8 | 111.5 |
| Mannitol | - | 115 | - | - | - | - | - |
| Primojel type A | 5.4 | 5.4 | 5.4 | 5.4 | 5.4 | 5.4 | 5.4 |
| Butylhydroxytoluene(BHT) | - | - | - | - | - | 0.27 | - |
| Citric acid | - | - | - | - | - | - | 3.5 |
| Meglumine | - | - | - | 0.781 | 3.125 | - | - |
| Magnesium stearate | 1.35 | 1.35 | 4.05 | 1.35 | 1.35 | 1.35 | 1.35 |
| Tablet weight (mg) | 135 | 135 | 135 | 135 | 135 | 135 | 135 |
| Compound | IClSM | IClRM | IClEt |
|---|---|---|---|
| Empirical formula | C27H37N2O5···Cl ··C8H8O3 | C27H37N2O5···Cl ··C8H8O3 | C27H37N2O5···Cl ··C2H6O |
| Formula wt. | 657.20 | 657.20 | 551.12 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic |
| Space group | P 212121 | P 212121 | P 21 |
| a (Å) | 7.4447(4) | 7.5398(2) | 5.4844(10) |
| b (Å) | 8.5027(4) | 8.6296(2) | 11.7967(10) |
| c (Å) | 52.159(2) | 51.5013(13) | 21.8297(10) |
| β (°) | 90 | 90 | 91.905(10) |
| V (Å3) | 3301.67(13) | 3350.96(8) | 1411.55(6) |
| Z | 4 | 4 | 2 |
| T (K) | 120 | 120 | 100 |
| ρcalc (g cm-3) | 1.322 | 1.303 | 1.297 |
| Radiation | CuKα (1.5418 Å) | CuKα (1.5418 Å) | CuKα (1.5418 Å) |
| μ (mm−1) | 1.477 | 1.456 | 1.565 |
| F(000) | 1400.0 | 1400.0 | 592.0 |
| Reflns collected | 12,420 | 11,567 | 20,662 |
| Indep. Reflns (Rint) | 5425 (0.057) | 5853 (0.035) | 5586 (0.083) |
| GOF | 0.962 | 1.039 | 1.160 |
| R1, wR2 [I>2σ(I)] | 0.045, 0.062 | 0.043, 0.056 | 0.049, 0.082 |
| R1, wR2 (all data) | 0.057, 0.067 | 0.046, 0.057 | 0.052, 0.084 |
| Res. el. dens. (e·Å−3) | 0.36, −0.32 | 0.44, −0.33 | 0.38, −0.34 |
| CCDC number | 1,479,092 | 1,479,091 | 1,479,090 |
| Stress Conditions | Form II | Form γ | Cocrystal | |||
|---|---|---|---|---|---|---|
| Solid Form, XRPD | Sum of Chem. Imp. [%] | Solid Form, XRPD | Sum of Chem. Imp. [%] | Solid Form, XRPD | Sum of Chem. Imp. [%] | |
| 3 days 80 °C | Form II + γ | 0.15 | Form γ | ≤0.05 | Cocrystal | ≤0.05 |
| 3 days 80 °C/75% RH | Form γ | 2.12 | Form γ | ≤0.05 | Cocrystal | ≤0.05 |
| 10 days 0% RH | Form II | 0.15 | Form γ | ≤0.05 | Cocrystal | ≤0.05 |
| 10 days 100% RH | Form β | 0.14 | Form β | ≤0.05 | Cocrystal | ≤0.05 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sládková, V.; Dammer, O.; Sedmak, G.; Skořepová, E.; Kratochvíl, B. Ivabradine Hydrochloride (S)-Mandelic Acid Co-Crystal: In Situ Preparation during Formulation. Crystals 2017, 7, 13. https://doi.org/10.3390/cryst7010013
Sládková V, Dammer O, Sedmak G, Skořepová E, Kratochvíl B. Ivabradine Hydrochloride (S)-Mandelic Acid Co-Crystal: In Situ Preparation during Formulation. Crystals. 2017; 7(1):13. https://doi.org/10.3390/cryst7010013
Chicago/Turabian StyleSládková, Veronika, Ondřej Dammer, Gregor Sedmak, Eliška Skořepová, and Bohumil Kratochvíl. 2017. "Ivabradine Hydrochloride (S)-Mandelic Acid Co-Crystal: In Situ Preparation during Formulation" Crystals 7, no. 1: 13. https://doi.org/10.3390/cryst7010013
APA StyleSládková, V., Dammer, O., Sedmak, G., Skořepová, E., & Kratochvíl, B. (2017). Ivabradine Hydrochloride (S)-Mandelic Acid Co-Crystal: In Situ Preparation during Formulation. Crystals, 7(1), 13. https://doi.org/10.3390/cryst7010013