
Aromatic C-Nitroso Compounds and Their Dimers: A Model for Probing the Reaction Mechanisms in Crystalline Molecular Solids


Abstract
:1. Introduction
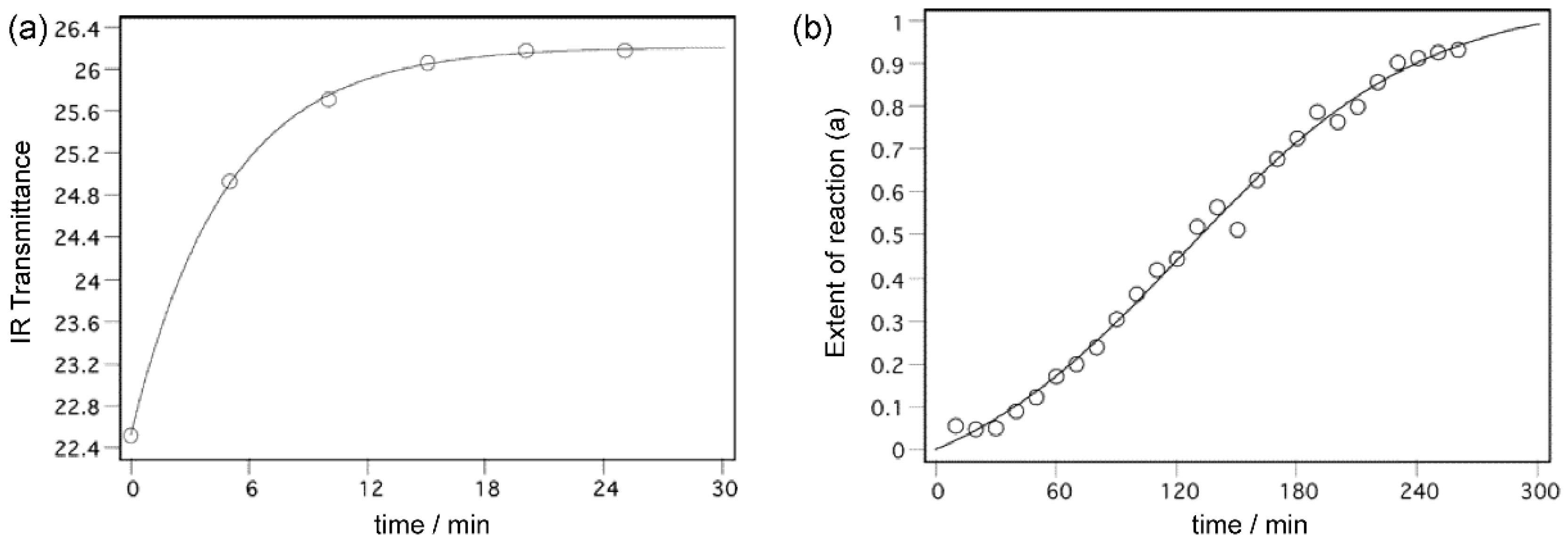
2. Experimental and Conceptual Model
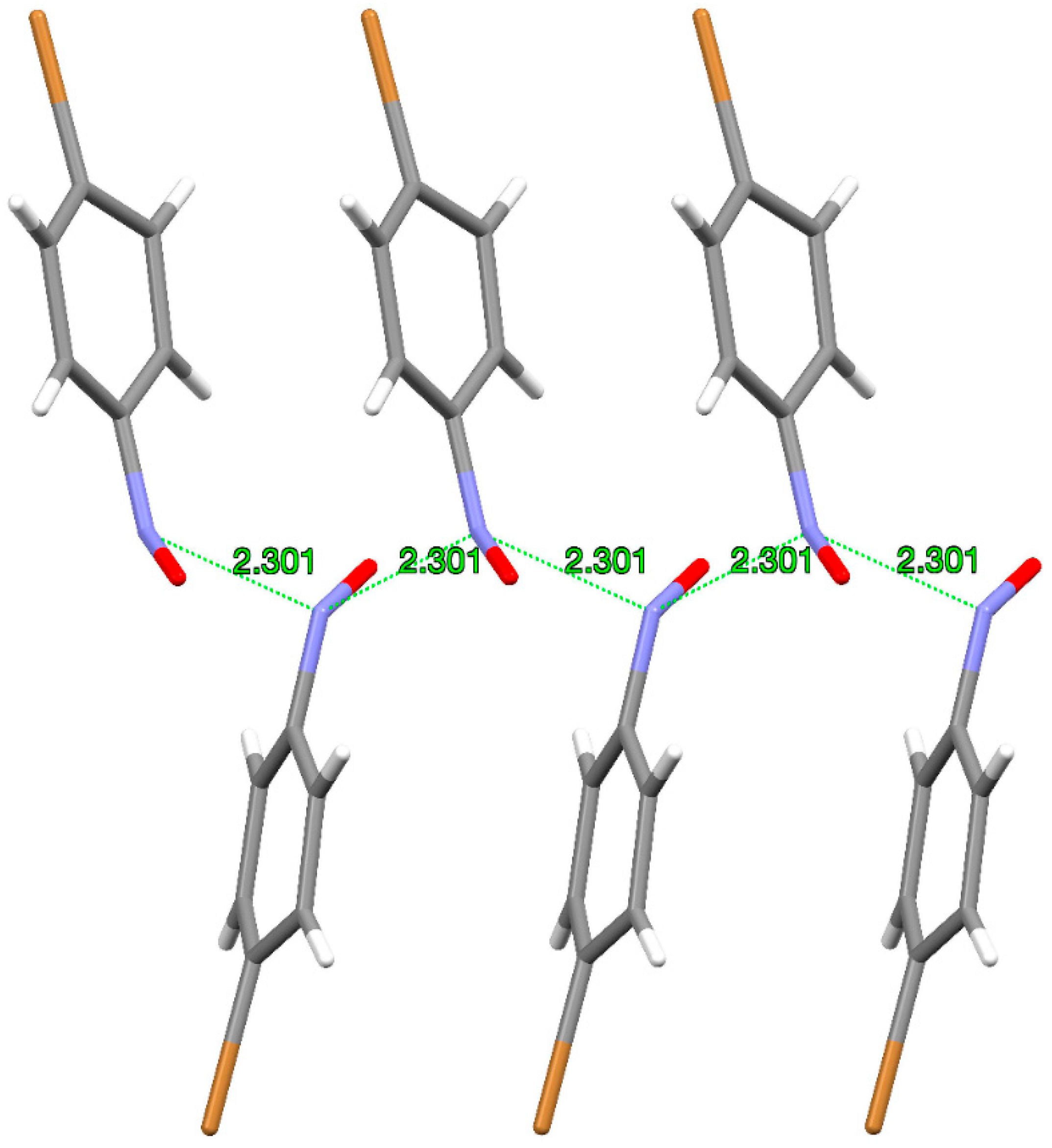
3. Dimerization Starting from the “Intimate Monomer Pair”
4. Dimerization Starting from the Van Der Waals Distant Monomers
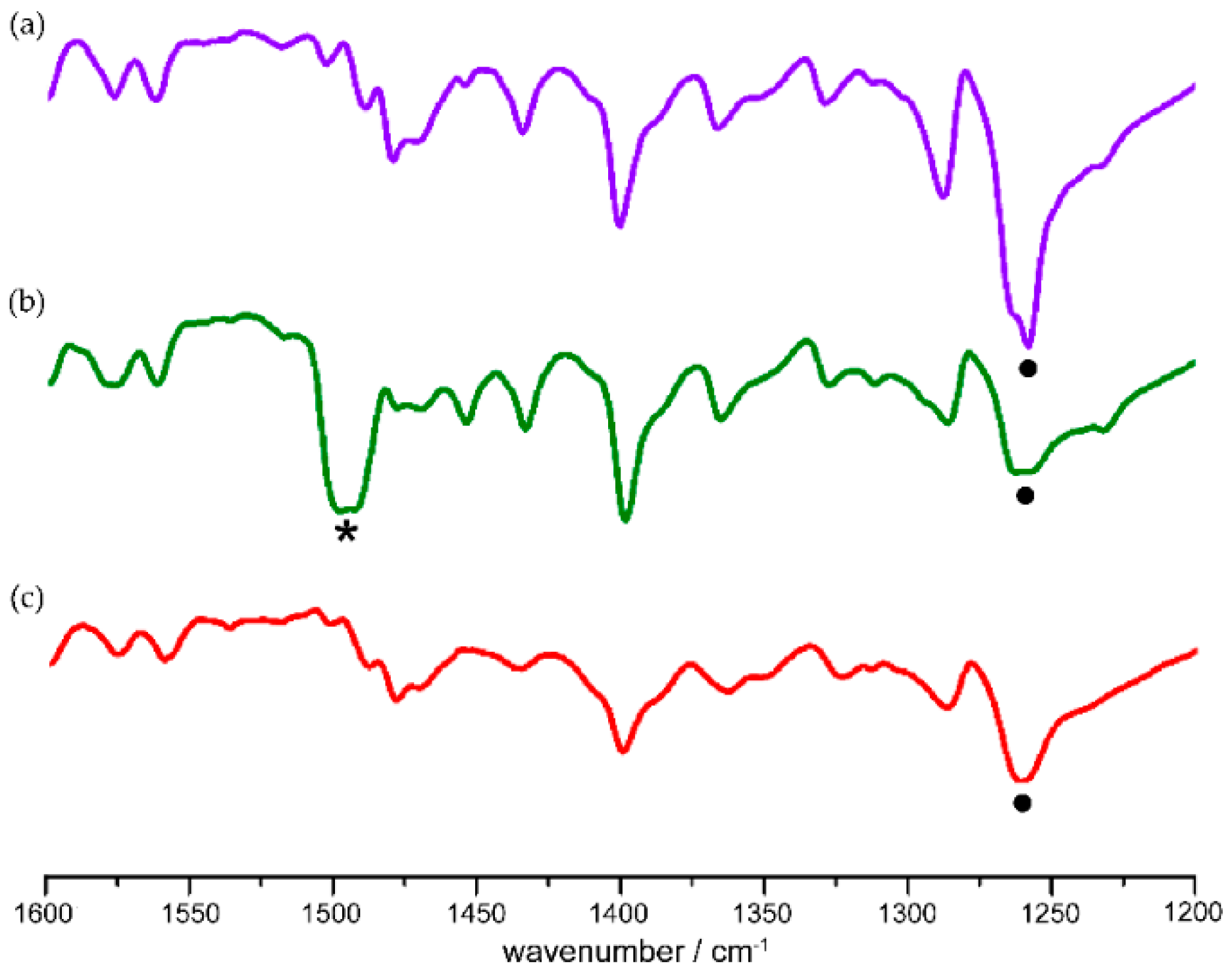
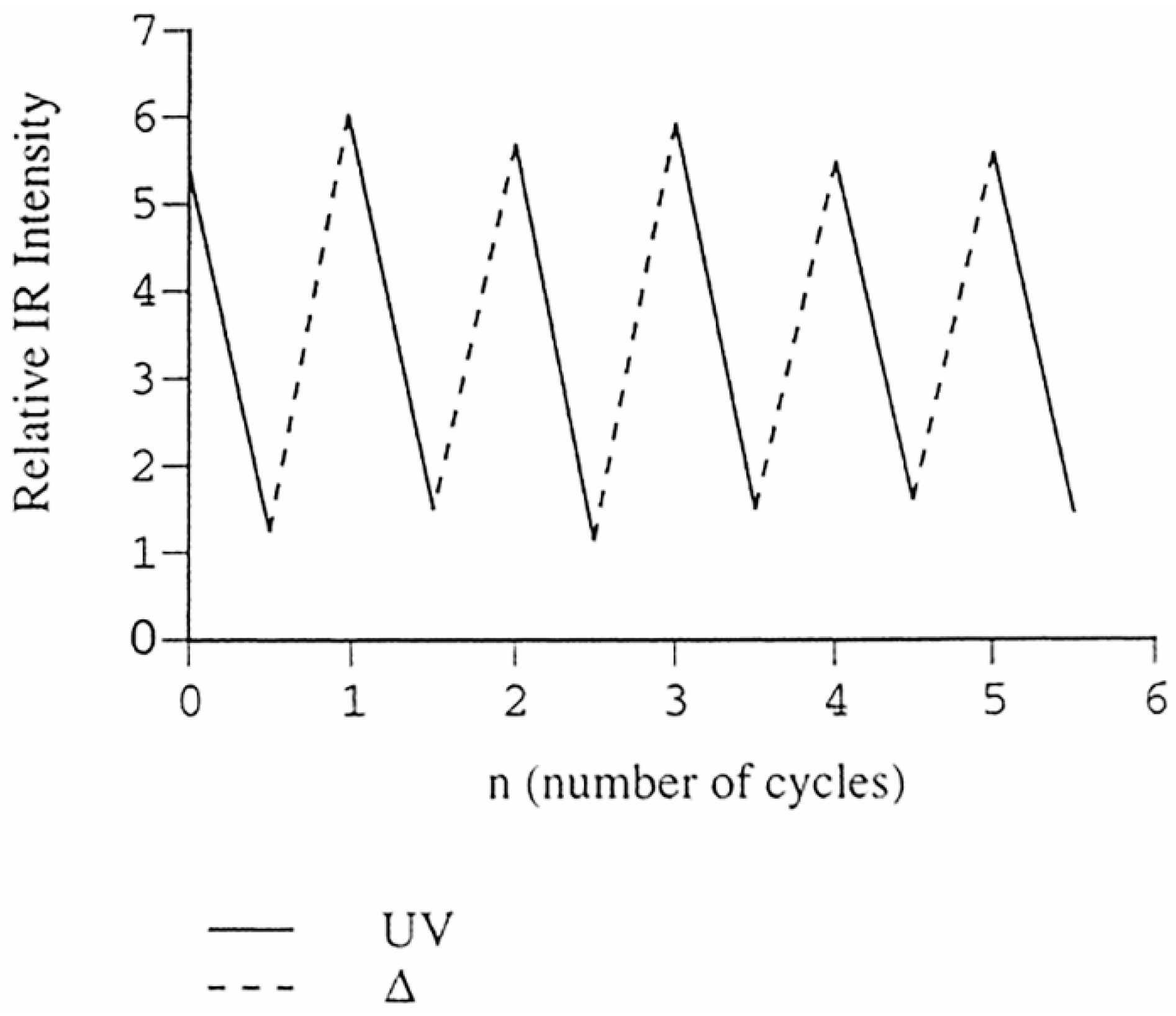
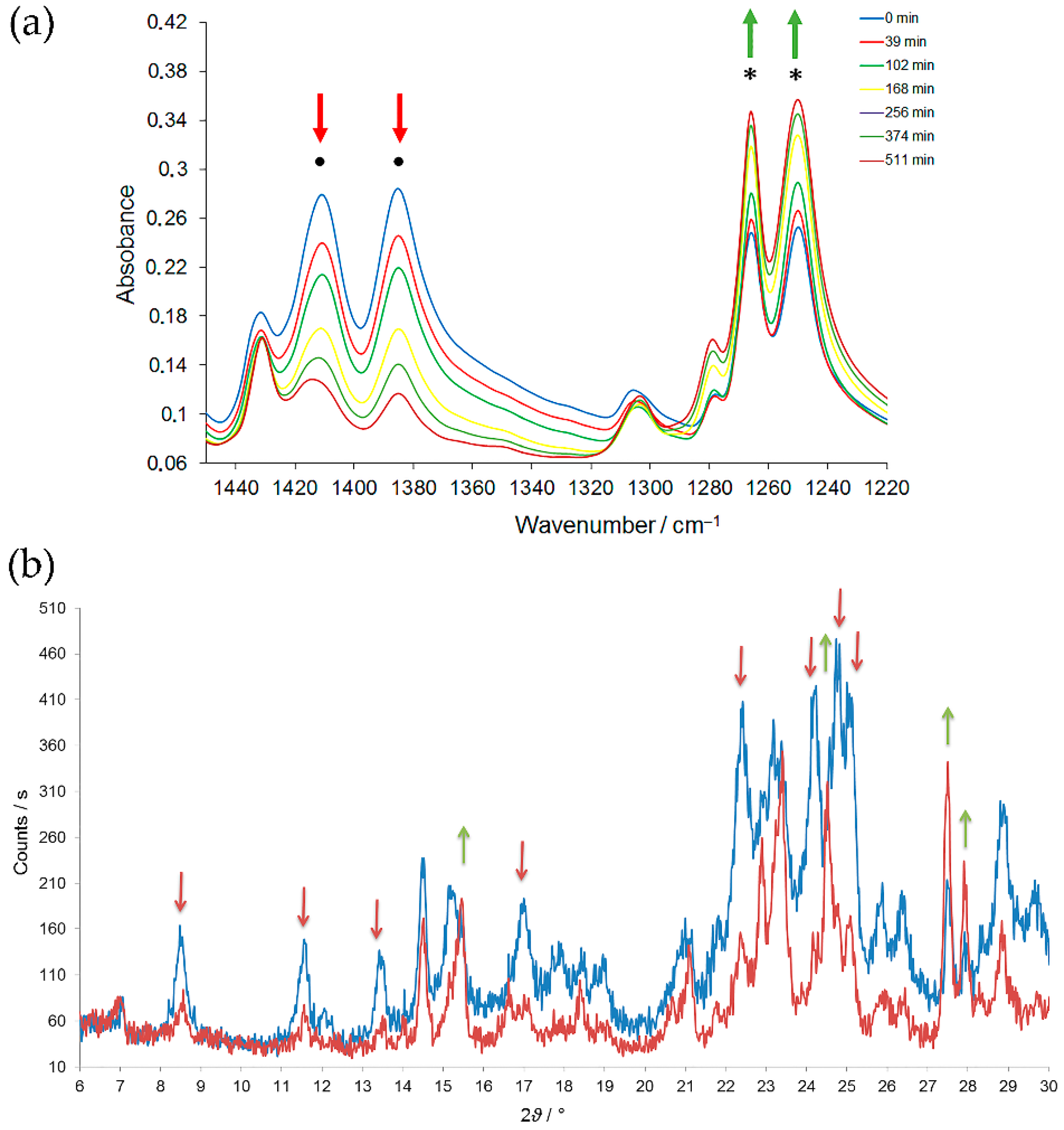
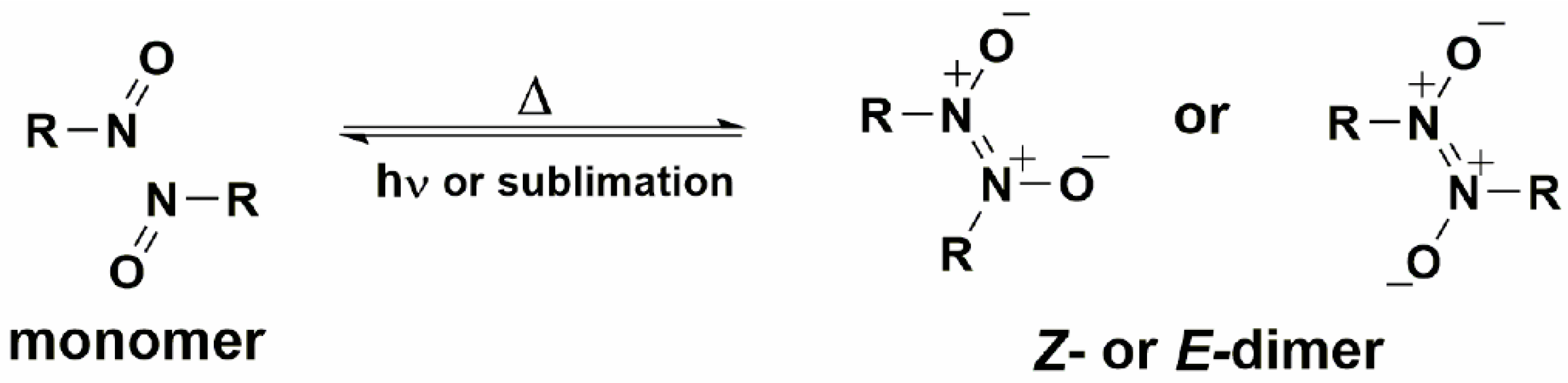
5. Solid-State Z-E Interconversion
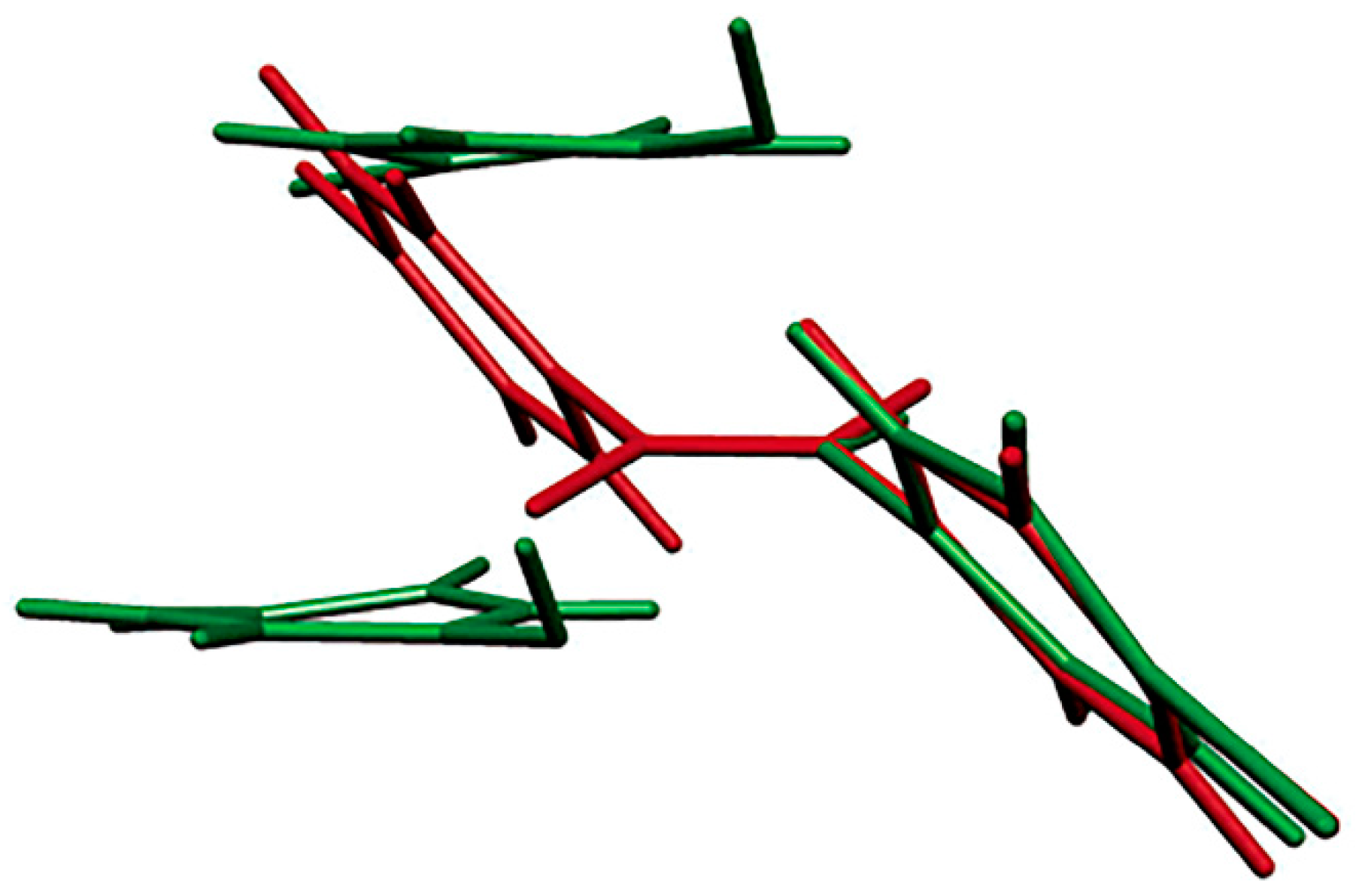
6. Surfaces and Formation of Crystal Defects
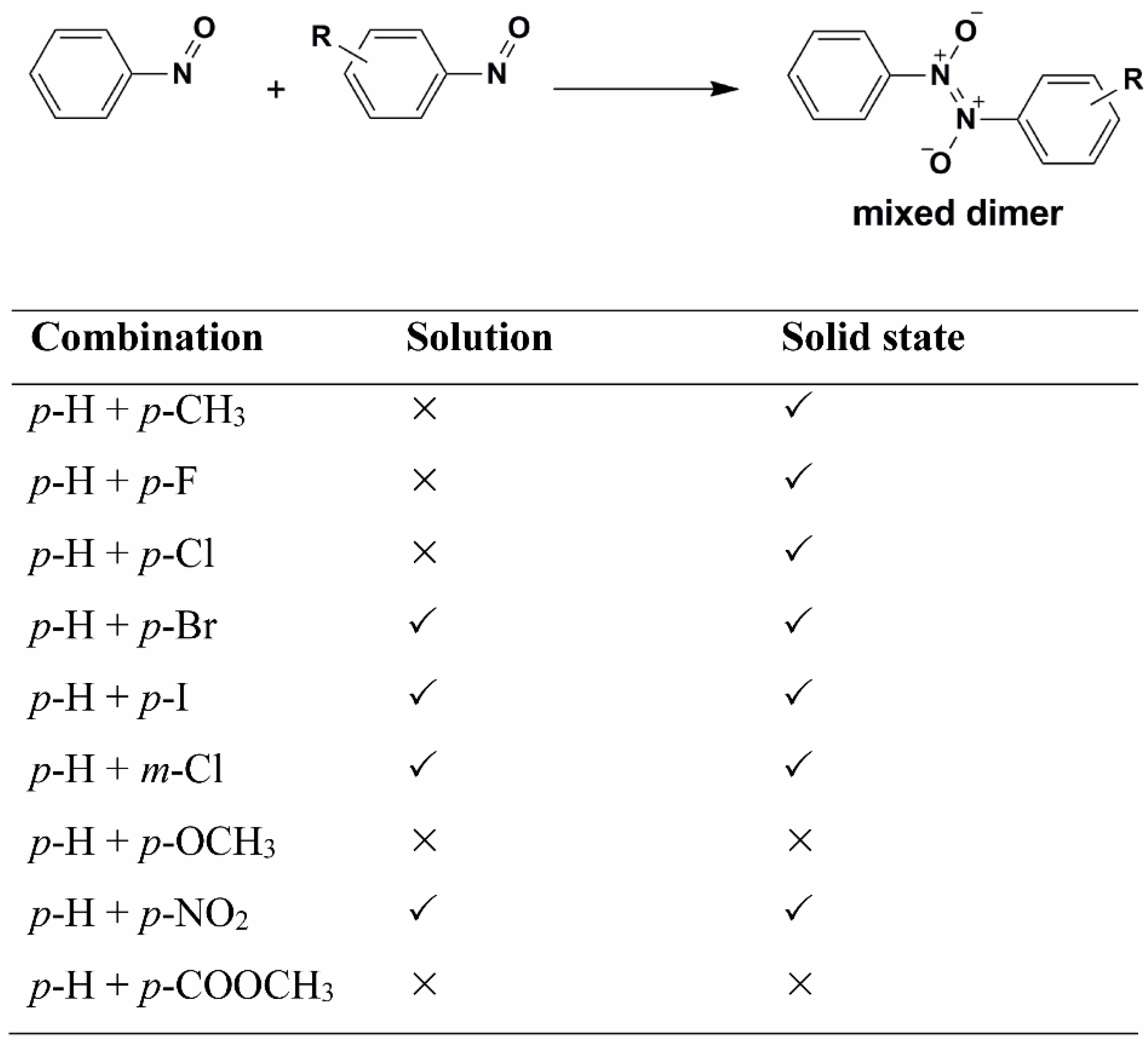
7. Metathesis-Like Reactions
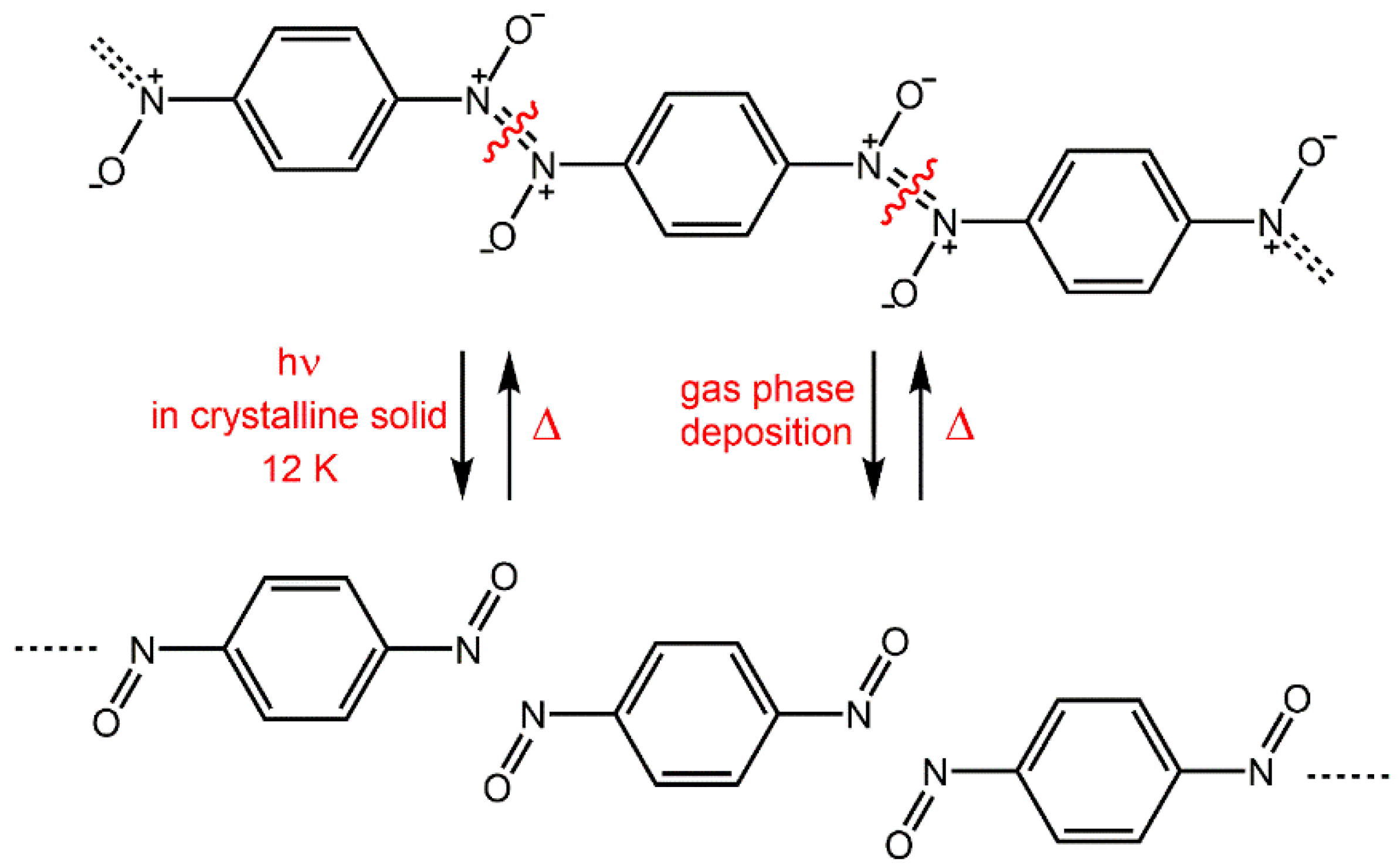
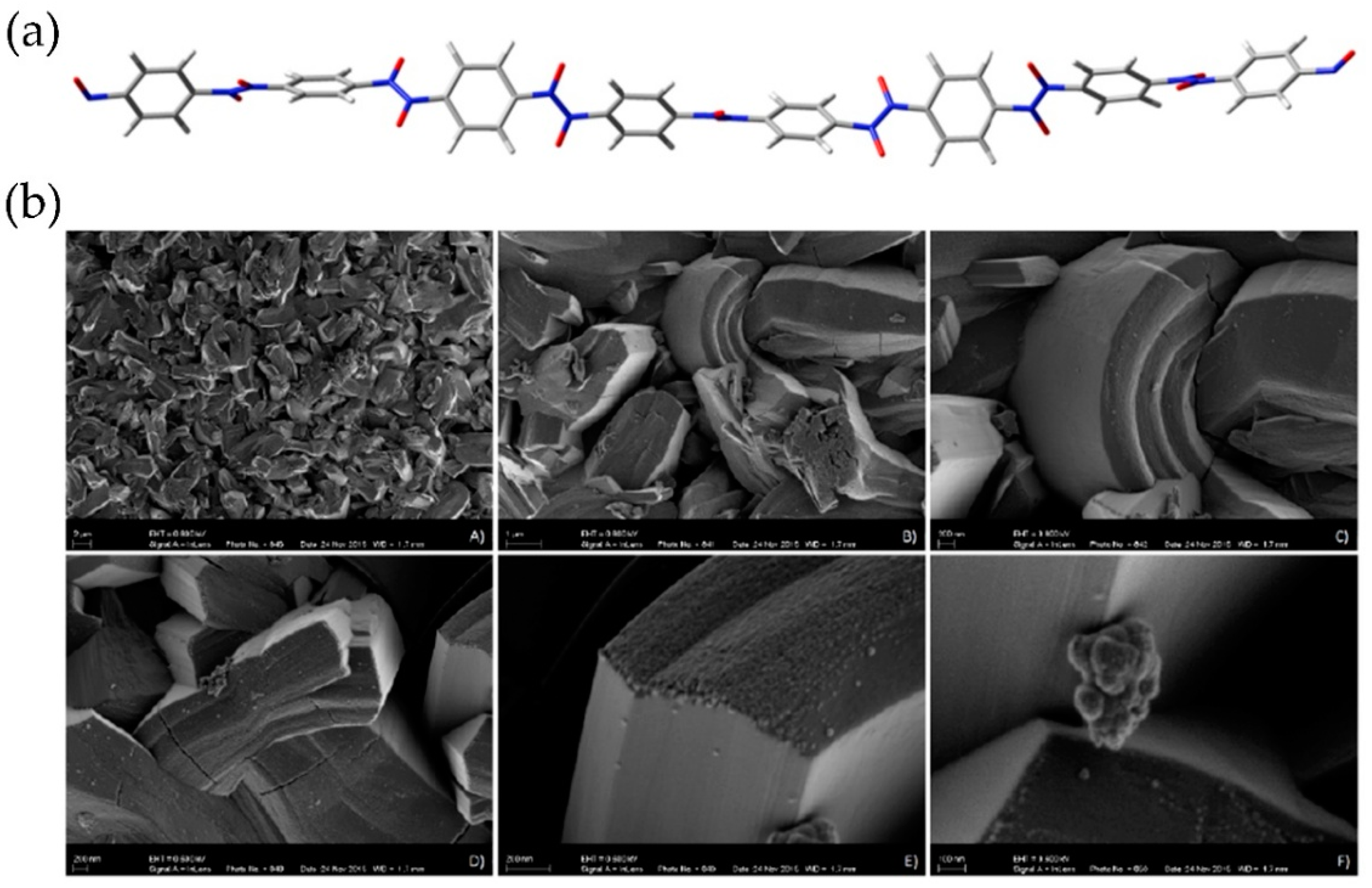
8. One-Dimensional Polymerization Reactions
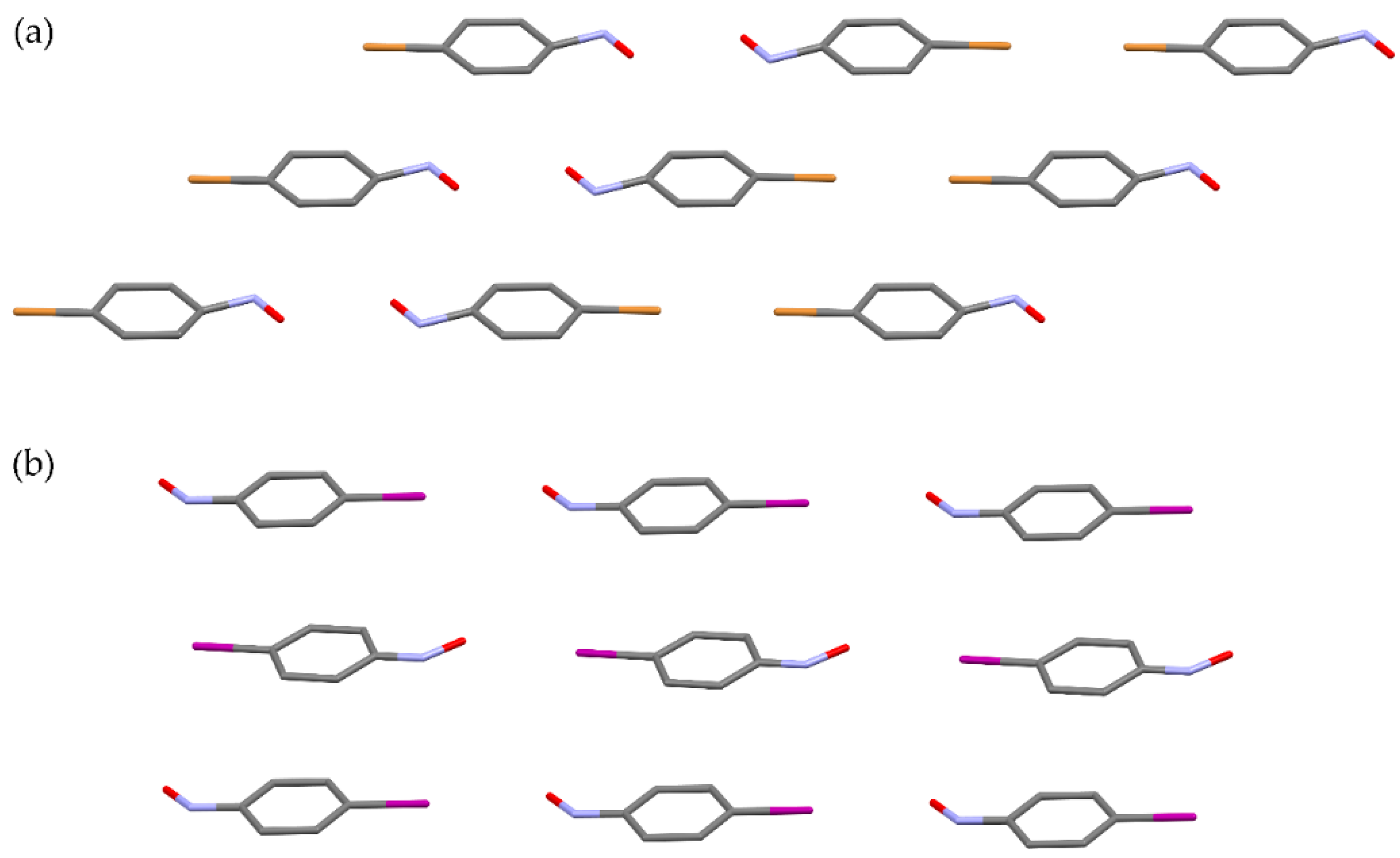
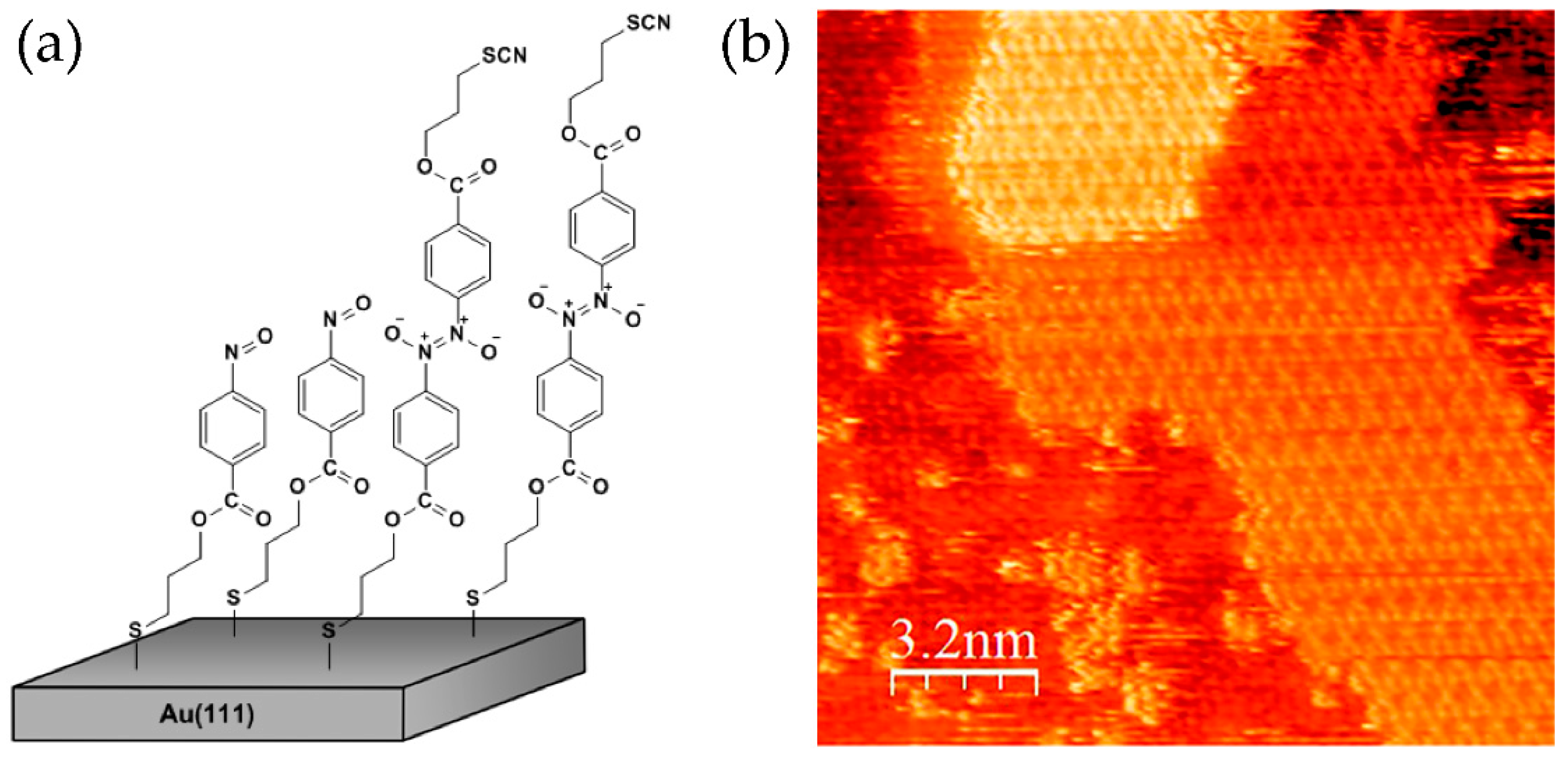
9. Self-Assembly and Dimerization on a Gold Surface
10. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- West, A.R. Solid State Chemistry and Its Applications, 2nd ed.; Wiley-Blackwell: Chichester, UK, 2014. [Google Scholar]
- Smart, L.E.; Moore, E.A. Solid State Chemistry: An Introduction, 4th ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Cohen, M.D.; Schmidt, G.M.J. 383. Topochemistry. Part I. A survey. J. Chem. Soc. 1964, 383, 1996–2000. [Google Scholar] [CrossRef]
- Kaupp, G. Solid-state molecular syntheses: Complete reactions without auxiliaries based on the new solid-state mechanism. CrystEngComm 2003, 5, 117–133. [Google Scholar] [CrossRef]
- Kaupp, G. Organic solid-state reactions with 100% yield. Top. Curr. Chem. 2005, 254, 95–183. [Google Scholar]
- Schmidt, G.M.J. Photodimerization in the solid state. Pure Appl. Chem. 1971, 27, 647–678. [Google Scholar] [CrossRef]
- Schmidt, G.M.J. Solid State Photochemistry; Verlag Chemie: Weinheim, NY, USA, 1976. [Google Scholar]
- Toda, F. Thermal and photochemical reactions in the solid state. Top. Curr. Chem. 2005, 254, 1–40. [Google Scholar]
- Bürgi, H.B. Structure correlation and chemistry. Acta Crystallogr. A 1998, 54, 873–885. [Google Scholar] [CrossRef]
- Bürgi, H.B.; Dunitz, J.D. From crystal statics to chemical dynamics. Acc. Chem. Res. 1983, 16, 153–161. [Google Scholar] [CrossRef]
- Dunitz, J.D. Phase transitions in molecular crystals from a chemical viewpoint. Pure Appl. Chem. 1991, 63, 177–185. [Google Scholar] [CrossRef]
- Paul, D.; Curtin, Y. Thermally induced organic reactions in the solid state. Acc. Chem. Res. 1973, 6, 217–225. [Google Scholar] [CrossRef]
- Vančik, H.; Šimunić-Mežnarić, V.; Meštrović, E.; Halasz, I. Nitrosobenzene Dimerizations as a model system for studying solid-state reaction mechanisms. J. Org. Chem. 2004, 69, 4829–4834. [Google Scholar] [CrossRef] [PubMed]
- Galwey, A.K.; Brown, M.E. Thermal Decomposition of Ionic Solids, Volume 86: Chemical Properties and Reactivities of Ionic Crystalline Phases (Studies in Physical and Theoretical Chemistry), 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Avrami, M. Kinetics of phase change. I General theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. II Transformation-Time relations for random distribution of nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Granulation, phase change, and microstructure kinetics of phase change. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Erofeev, B.V. Generalized equation of chemical kinetics and its application in reactions involving solids. Compt. Rend. Acad. Sci. USSR 1946, 52, 511–514. [Google Scholar]
- Finney, E.E.; Finke, R.G. Nanocluster nucleation and growth kinetic and mechanistic studies: A review emphasizing transition-metal nanoclusters. J. Colloid Interface Sci. 2008, 317, 351–374. [Google Scholar] [CrossRef] [PubMed]
- Finney, E.E.; Finke, R.G. Is there a minimal chemical mechanism underlying classical Avrami-Erofeev treatments of phase-transformation kinetic data? Chem. Mater. 2009, 21, 4692–4705. [Google Scholar] [CrossRef]
- Finney, E.E.; Finke, R.G. Fitting and interpreting transition-metal nanocluster formation and other sigmoidal-appearing kinetic data: A more thorough testing of dispersive kinetic vs chemical-mechanism-based equations and treatments for 4-step type kinetic data. Chem. Mater. 2009, 21, 4468–4479. [Google Scholar] [CrossRef]
- Watzky, M.A.; Finke, R.G. Transition metal nanocluster formation kinetic and mechanistic studies. A new mechanism when hydrogen is the reductant: Slow, continuous nucleation and fast autocatalytic surface growth. J. Am. Chem. Soc. 1997, 119, 10382–10400. [Google Scholar] [CrossRef]
- Brown, M.E. Steps in a minefield. J. Therm. Anal. 1997, 49, 17–32. [Google Scholar] [CrossRef]
- Galwey, A.K. What can we learn about the mechanisms of thermal decompositions of solids from kinetic measurements? J. Therm. Anal. Calorim. 2008, 92, 967–983. [Google Scholar] [CrossRef]
- Vančik, H. Aromatic C-Nitroso Compounds; Springer: New York, NY, USA, 2013. [Google Scholar]
- Halasz, I.; Meštrović, E.; Čičak, H.; Mihalić, Z.; Vančik, H. Solid-State reaction mechanisms in monomer–dimer interconversions of p-Bromonitrosobenzene. single-crystal-to-single-crystal photodissociation and formation of new non-van der Waals close contacts. J. Org. Chem. 2005, 70, 8461–8467. [Google Scholar] [CrossRef] [PubMed]
- Halasz, I.; Vančik, H. Surface nucleation in solid-state dimerisation of nitrosobenzenes promoted by sublimation. CrystEngComm 2011, 13, 4307–4310. [Google Scholar] [CrossRef]
- Maganjić, A.; Šolić, I.; Milovac, S.; Halasz, I.; Biljan, I.; Vančik, H. Mechanochemically induced cross-dimerizations of nitrosobenzenes. Kinetics and solid-state isotope effects. J. Phys. Org. Chem. 2014, 27, 177–182. [Google Scholar] [CrossRef]
- Varga, K.; Vančik, H. Topochemical effect in thermal E-Z isomerization of azodioxides in solid state. J. Phys. Org. Chem. 2016, 29, 214–220. [Google Scholar] [CrossRef]
- Varga, K.; Volarić, J.; Vančik, H. Crystal disordering and organic solid-state reactions. CrystEngComm 2015, 17, 1434–1438. [Google Scholar] [CrossRef]
- Knežević, A.; Medančić, T.; Milovac, S.; Biljan, I.; Halasz, I.; Vančik, H. Photothermal reactions of nitrosobenzene and halonitrosobenzenes in solid-state. Croat. Chem. Acta 2011, 84, 21–24. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Gowenlock, B.G.; Orrell, K.G. Structural investigations of C-nitrosobenzenes. Part 1. Solution state 1H NMR studies. J. Chem. Soc. Perkin Trans. 2 1997, 2201–2206. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Gowenlock, B.G.; Orrell, K.G. Structural investigations of C-nitrosobenzenes. Part 2.1 NMR studies of monomer-dimer equilibria including restricted nitroso group rotation in monomers. J. Chem. Soc. Perkin Trans. 2 1998, 797–804. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Gowenlock, B.G.; Orrell, K.G.; Šik, V. Dynamic NMR study of the factors governing nitroso group rotation in p-nitrosoanilines in the solution and solid states. Magn. Reson. Chem. 1995, 33, 561–569. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Gowenlock, B.G.; Orrell, K.G.; Šik, V.; Hibbs, D.E.; Hursthouse, M.B.; Malik, A.K.M. 4-Iodonitrosobenzene. Structural and spectroscopic studies of the monomeric solid and of previously unreported dimers. J. Chem. Soc. Perkin Trans. 2 1996, 191–197. [Google Scholar] [CrossRef]
- Gowenlock, B.G.; Maidment, M.J.; Orrell, K.G.; Šik, V.; Mele, G.; Vasapollo, G.; Hursthouse, M.B.; Abdul Malik, K.M. The solid- and solution-state structures of 2-nitrosopyridine and its 3- and 4-methyl derivatives. J. Chem. Soc. Perkin Trans. 2 2000, 2280–2286. [Google Scholar] [CrossRef]
- Gowenlock, B.G.; Richter-Addo, G.B. Dinitroso and polynitroso compounds. Chem. Soc. Rev. 2005, 34, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, D.; Maris, T.; Wuest, J.D. Constructing monocrystalline covalent organic networks by polymerization. Nat. Chem. 2013, 5, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, D.; Wuest, J.D. Dimerization of aromatic C-nitroso compounds. Chem. Rev. 2016, 116, 258–286. [Google Scholar] [CrossRef] [PubMed]
- Vančik, H.; Šimunić-Mežnarić, V.; Meštrović, E.; Milovac, S.; Majerski, K.; Veljković, J. Solid state photochromism and thermochromism in nitroso monomer-dimer equilibrium. J. Phys. Chem. B 2002, 106, 1576–1580. [Google Scholar] [CrossRef]
- Glaser, R.; Murmann, R.K.; Barnes, C.L. Why do nitroso compounds dimerize while their oxime tautomers do not? A structural study of the trans-dimer of 2-chloro-2-methyl-3-nitrosobutane and higher level ab initio study of thermodynamic stabilities and electronic structures of isomers of diazene dioxides. J. Org. Chem. 1996, 61, 1047–1058. [Google Scholar]
- Novak, I. Computational thermochemistry of C-nitroso compounds. Struct. Chem. 2016, 27, 1395–1401. [Google Scholar] [CrossRef]
- Rassat, A.; Rey, P. Nitroxides: Photochemical synthesis of trimethylisoquinuclidine N-Oxyl. J. Chem. Soc. D 1971, 1161–1162. [Google Scholar] [CrossRef]
- Ullman, E.; Call, L.; Tseng, S.S. Stable free radicals. XII. Direct and sensitized nitronyl nitroxide photochemistry in aprotic solvent. J. Am. Chem. Soc. 1973, 95, 1677–1679. [Google Scholar] [CrossRef]
- Ullman, E.F.; Singh, P. 3,3,4,4-Tetramethyl-1,2-diazetine 1,2-dioxide, a useful low-energy triplet quencher. J. Am. Chem. Soc. 1972, 94, 5077–5078. [Google Scholar] [CrossRef]
- Dunkin, I.R.; Lynch, M.A.; Boulton, A.J.; Henderson, N. 1,2-Dinitrosobenzene in argon matrices at 14 K. J. Chem. Soc. Chem. Commun. 1991, 1178–1179. [Google Scholar] [CrossRef]
- Azoulay, M.; Fischer, E. Low-temperature proton nuclear magnetic resonance and ultraviolet absorption spectra and photochemistry of the system nitrosobenzene-azodioxybenzene and its methyl derivatives. J. Chem. Soc. Perkin Trans. 2 1982, 637–642. [Google Scholar] [CrossRef]
- Hoffmann, R.; Gleiter, R.; Mallory, F.B. Non-least-motion potential surfaces. Dimerization of methylenes and nitroso compounds. J. Am. Chem. Soc. 1970, 92, 1460–1466. [Google Scholar] [CrossRef]
- Bibulić, P.; Rončević, I.; Varga, K.; Mihalić, Z.; Vančik, H. Structure and topochemistry of azodioxide oligomers in solid state. J. Mol. Struct. 2016, 1104, 85–90. [Google Scholar] [CrossRef]
- Bibulić, P.; Rončević, I.; Bermanec, V.; Vančik, H. Polymerization of 1,4-dinitrosobenzene: Kinetics and submicrocrystal structure. Croat. Chem. Acta 2017, in press. [Google Scholar]
- Gowenlock, B.G.; Trotman, J. Geometrical isomerism of dimeric nitrosomethane. J. Chem. Soc. 1955, 4190–4196. [Google Scholar] [CrossRef]
- Chaudhry, A.U.; Gowenlock, B.G. Thermal cis→trans isomerization of two dimeric nitrosoalkanes. J. Chem. Soc. B 1968, 1083–1084. [Google Scholar] [CrossRef]
- Wajer, T.A.J.; de Boer, T.J. Chemistry of small ring compounds. Part 16: Kinetics and mechanism of the isomerization of some 1,1-cyclopropanedicarboxylic acids in water and in aqueous sulfuric acid. Recl. Trav. Chim. Pays-Bas 1972, 91, 657–666. [Google Scholar]
- Minato, T.; Yamabe, S.; Oda, H. A theoretical study on the cis/trans-isomerization of azodioxymethane. Can. J. Chem. 1982, 60, 2740–2748. [Google Scholar] [CrossRef]
- Orrell, K.G.; Šik, V.; Stephenson, D. Study of the monomer-dimer equilibrium of nitrosobenzene using multinuclear one- and two-dimensional NMR techniques. Magn. Reson. Chem. 1987, 25, 1007–1011. [Google Scholar] [CrossRef]
- Mnyukh, Y. Mechanism and kinetics of phase transitions and other reactions in solids. AJCMP 2013, 3, 89–103. [Google Scholar]
- Rao, C.N.R.; Gopalakrishnan, J. New Directions in Solid State Chemistry, 2nd ed.; Cambridge University Press: Cambridge, UK, 1997. [Google Scholar]
- Shalaev, E.; Shalaeva, M.; Zografi, G. The effect of disorder on the chemical reactivity of an organic solid, tetraglycine methyl ester: Change of the reaction mechanism. J. Pharm. Sci. 2002, 91, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Stowell, J.G.; Cao, W.; Morris, K.R.; Byrn, S.R.; Carvajal, M.T. Effect of milling and compression on the solid-state maillard reaction. J. Pharm. Sci. 2005, 94, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.S. An X-ray examination of the crystal structure of p-iodonitrosobenzene. J. Chem. Soc. 1956, 2841–2845. [Google Scholar] [CrossRef]
- Halasz, I.; Biljan, I.; Novak, P.; Meštrović, E.; Plavec, J.; Mali, G.; Smrečki, V.; Vančik, H. Cross-dimerization of nitrosobenzenes in solution and in solid state. J. Mol. Struct. 2009, 918, 19–25. [Google Scholar] [CrossRef]
- Biljan, I.; Cvjetojević, G.; Smrečki, V.; Novak, P.; Mali, G.; Plavec, J.; Babić, D.; Mihalić, Z.; Vančik, H. Nitrosobenzene cross-dimerization: Structural selectivity in solution and in solid state. J. Mol. Struct. 2010, 979, 22–26. [Google Scholar] [CrossRef]
- Hacker, N.P. Investigation of the polymerization of 1,4-dinitrosobenzene by low-temperature infrared and UV absorption spectroscopy. Macromolecules 1993, 26, 5937–5942. [Google Scholar] [CrossRef]
- Biljan, I.; Kralj, M.; Mišić Radić, T.; Svetličić, V.; Vančik, H. Dimerization of nitrosobenzene derivatives on an Au(111) Surface. J. Phys. Chem. C 2011, 115, 20267–20273. [Google Scholar] [CrossRef]
- Biljan, I.; Medančić, T.; Kralj, M.; Mišić Radić, T.; Svetličić, V.; Vančik, H. Nitrosoarene dimerization on two- and three-dimensional gold surfaces. Croat. Chem. Acta 2013, 86, 83–94. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T/K | 104 k/s−1 | Ea/kJ mol−1 |
|---|---|---|
| 293 | 1.78 ± 0.37 a | 59.27 ± 0.44 |
| 298 | 3.33 ± 0.15 | |
| 303 | 4.41 ± 0.10 | |
| 308 | 7.51 ± 0.63 | |
| 313 | 8.21 ± 0.31 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biljan, I.; Vančik, H. Aromatic C-Nitroso Compounds and Their Dimers: A Model for Probing the Reaction Mechanisms in Crystalline Molecular Solids. Crystals 2017, 7, 376. https://doi.org/10.3390/cryst7120376
Biljan I, Vančik H. Aromatic C-Nitroso Compounds and Their Dimers: A Model for Probing the Reaction Mechanisms in Crystalline Molecular Solids. Crystals. 2017; 7(12):376. https://doi.org/10.3390/cryst7120376
Chicago/Turabian StyleBiljan, Ivana, and Hrvoj Vančik. 2017. "Aromatic C-Nitroso Compounds and Their Dimers: A Model for Probing the Reaction Mechanisms in Crystalline Molecular Solids" Crystals 7, no. 12: 376. https://doi.org/10.3390/cryst7120376
APA StyleBiljan, I., & Vančik, H. (2017). Aromatic C-Nitroso Compounds and Their Dimers: A Model for Probing the Reaction Mechanisms in Crystalline Molecular Solids. Crystals, 7(12), 376. https://doi.org/10.3390/cryst7120376