
Molecular Dynamics Simulations of Hydroxyapatite Nanopores in Contact with Electrolyte Solutions: The Effect of Nanoconfinement and Solvated Ions on the Surface Reactivity and the Structural, Dynamical, and Vibrational Properties of Water

 ,
,  ,
,
Abstract
:1. Introduction
2. Computational Methods
2.1. Simulation Details
2.2. Interatomic Potential Model
2.3. Simulation Protocol
2.4. Analysis of the Simulation Data
3. Results and Discussion
3.1. Structural, Dynamical, and Vibrational Properties of Water
3.2. Kinetics of Water Exchange at the HAP-Solution Interface
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Loste, E.; Park, R.J.; Warren, J.; Meldrum, F.C. Precipitation of calcium carbonate in confinement. Adv. Funct. Mater. 2004, 14, 1211–1220. [Google Scholar] [CrossRef]
- Meldrum, F.C.; Cölfen, H. Controlling mineral morphologies and structures in biological and synthetic systems. Chem. Rev. 2003, 108, 4332–4432. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Christenson, H.K.; Meldrum, F.C. Confinement Increases the Lifetimes of Hydroxyapatite Precursors. Chem. Mater. 2014, 26, 5830–5838. [Google Scholar] [CrossRef]
- Schenk, A.S.; Albarracin, E.; Kim, Y.-Y.; Ihli, J.; Meldrum, F.C. Confinement stabilises single crystal vaterite rods. Chem. Commun. 2014, 50, 4729–4732. [Google Scholar] [CrossRef] [PubMed]
- Verch, A.; Côté, A.S.; Darkins, R.; Kim, Y.-Y.; van de Locht, R.; Meldrum, F.C.; Duffy, D.; Kröger, R. Correlation between Anisotropy and Lattice Distortions in Single Crystal Calcite Nanowires Grown in Confinement. Small 2014, 10, 2697–2702. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Feldman, J.; Lee, S.S. Nanoconfined Crystallization of MAPbI3 to Probe Crystal Evolution and Stability. Cryst. Growth Des. 2016, 16, 4744–4751. [Google Scholar] [CrossRef]
- Jiang, Q.; Ward, M.D. Crystallization under nanoscale confinement. Chem. Soc. Rev. 2014, 43, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Reeves, N.J.; Mann, S. Photoelectron spectroscopic studies of the silicon pseudohalides: Relationship between geometrical and electronic structure. J. Chem. Soc. Faraday Trans. 1991, 87, 3875–3880. [Google Scholar] [CrossRef]
- Didymus, J.M.; Oliver, P.; Mann, S.; de Vries, A.L.; Hauschka, P.V.; Westbroek, P. Influence of Low-Molecular-Weight and Macromolecular Organic Additives on the Morphology of Calcium Carbonate. J. Chem. Soc. Faraday Trans. 1993, 89, 2891–2900. [Google Scholar] [CrossRef]
- Lahav, M.; Leiserowitz, L. The effect of solvent on crystal growth and morphology. Chem. Eng. Sci. 2001, 56, 2245–2253. [Google Scholar] [CrossRef]
- De Yoreo, J.J.; Vekilov, P.G. Principles of crystal nucleation and grow. Rev. Mineral. Geochem. 2003, 54, 57–93. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, C.; Benning, L.G. Control of Crystal Nucleation and Growth by Additives. Elements 2013, 9, 203–209. [Google Scholar] [CrossRef]
- Hamilton, B.D.; Ha, J.M.; Hillmyer, M.A.; Ward, M.D. Manipulating crystal growth and polymorphism by confinement in nanoscale crystallization chambers. Acc. Chem. Res. 2012, 45, 414–423. [Google Scholar] [CrossRef]
- Mann, S.; Archibald, D.D.; Didymus, J.M.; Douglas, T.; Heywood, B.R.; Meldrum, F.C.; Reeves, N.J. Crystallization at Inorganic-organic Interfaces: Biominerals and Biomimetic Synthesis. Science 1993, 261, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Elhadj, S.; de Yoreo, J.J.; Hoyer, J.R.; Dove, P.M. Role of molecular charge and hydrophilicity in regulating the kinetics of crystal growth. Proc. Natl. Acad. Sci. USA 2006, 103, 19237–19242. [Google Scholar] [CrossRef] [PubMed]
- Chun-Long, C.; Jiahui, Q.; Zuckermann, R.N.; de Yoreo, J.J. Engineered Biomimetic Polymers as Tunable Agents for Controlling CaCO3 Mineralization. J. Am. Chem. Soc. 2011, 133, 5214–5217. [Google Scholar]
- Kowacz, M.; Putnis, A. The effect of specific background electrolytes on water structure and solute hydration: Consequences for crystal dissolution and growth. Geochim. Cosmochim. Acta 2008, 72, 4476–4487. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Kowacz, M.; Putnis, C.V.; Putnis, A. The role of background electrolytes on the kinetics and mechanism of calcite dissolution. Geochim. Cosmochim. Acta 2010, 74, 1256–1267. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Urosevic, M.; Putnis, C.V.; Rodriguez-Navarro, C.; Cardell, C.; Putnis, A. Ion-specific effects on the kinetics of mineral dissolution. Chem. Geol. 2011, 281, 364–371. [Google Scholar] [CrossRef]
- Weaver, M.L.; Qiu, S.R.; Hoyer, J.R.; Casey, W.H.; Nancollas, G.H.; de Yoreo, J.J. Inhibition of calcium oxalate monohydrate growth by citrate and the effect of the background electrolyte. J. Cryst. Growth 2007, 306, 135–145. [Google Scholar] [CrossRef]
- Lowenstam, H.A.; Weiner, S. On Biomineralization; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Narasaraju, T.S.B.; Phebe, D.E. Some physico-chemical aspects of hydroxylapatite. J. Mater. Sci. 1996, 31, 1. [Google Scholar] [CrossRef]
- Knothe Tate, M.L. “Whither flows the fluid in bone?” An osteocyte’s perspective. J. Biomech. 2003, 36, 1409–1424. [Google Scholar] [CrossRef]
- Cowin, S.C.; Gailani, G.; Benalla, M. Hierarchical poroelasticity: Movement of interstitial fluid between porosity levels in bones. Philos. Trans. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 2009, 367, 3401–3444. [Google Scholar] [CrossRef] [PubMed]
- Wehrli, F.W.; Fernandez-Seara, M.A. Nuclear Magnetic Resonance Studies of Bone Water. Ann. Biomed. Eng. 2005, 33, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, L.; Fritton, S.P.; Gailani, G.; Benalla, M.; Cowin, S.C. Advances in assessment of bone porosity, permeability and interstitial fluid flow. J. Biomech. 2013, 46, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Myerson, A.S.; Trout, B.L. Nucleation from Solution. Science 2013, 341, 855–856. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.E. Electrolyte crystal growth mechanisms. J. Cryst. Growth 1984, 67, 289–310. [Google Scholar] [CrossRef]
- Piana, S.; Jones, F.; Gale, J.D. Assisted desolvation as a key kinetic step for crystal growth. J. Am. Chem. Soc. 2006, 128, 13568–13574. [Google Scholar] [CrossRef] [PubMed]
- Pokrovsky, O.S.; Schott, J. Surface Chemistry and Dissolution Kinetics of Divalent Metal Carbonates. Environ. Sci. Technol. 2002, 36, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, N.Y.; Brown, P.W. Computer simulation of stoichiometric hydroxyapatite: Structure and substitutions. J. Phys. Chem. Solids 2007, 68, 431–437. [Google Scholar] [CrossRef]
- De Leeuw, N.H. Computer simulations of structures and properties of the biomaterial hydroxyapatite. J. Mater. Chem. 2010, 20, 5376–5389. [Google Scholar] [CrossRef]
- Zahn, D.; Hochrein, O. Computational study of interfaces between hydroxyapatite and water. Phys. Chem. Chem. Phys. 2003, 5, 4004–4007. [Google Scholar] [CrossRef]
- Habraken, W.J.E.M.; Tao, J.; Brylka, L.J.; Friedrich, H.; Bertinetti, L.; Schenk, A.S.; Verch, A.; Dmitrovic, V.; Bomans, P.H.H.; Frederik, P.M.; et al. Ion-association complexes unite classical and non-classical theories for the biomimetic nucleation of calcium phosphate. Nat. Commun. 2013, 4, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, G.; Terranova, U.; de Leeuw, N.H. Calcium Phosphate Prenucleation Complexes in Water by Means of ab Initio Molecular Dynamics Simulations. Cryst. Growth Des. 2016, 16, 3353–3358. [Google Scholar] [CrossRef]
- Orgel, J.P.R.O.; Irving, T.C.; Miller, A.; Wess, T.J. Microfibrillar structure of type I collagen in situ. Proc. Natl. Acad. Sci. USA 2006, 103, 9001–9005. [Google Scholar] [CrossRef] [PubMed]
- Cantaert, B.; Beniash, E.; Meldrum, F.C. Nanoscale Confinement Controls the Crystallization of Calcium Phosphate: Relevance to Bone Formation. Chem. Eur. J. 2013, 19, 14918–14924. [Google Scholar] [CrossRef] [PubMed]
- Todorov, I.T.; Smith, W.; Trachenko, K.; Dove, M.T. DL_POLY_3: New dimensions in molecular dynamics simulations via massive parallelism. J. Mater. Chem. 2006, 16, 1911–1918. [Google Scholar] [CrossRef]
- Melchionna, S.; Ciccotti, G.; Holian, B.L. Hoover NPT dynamics for systems varying in shape and size. Mol. Phys. 1993, 78, 533–544. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef]
- De Leeuw, N.H. Resisting the Onset of Hydroxyapatite Dissolution through the Incorporation of Fluoride. J. Phys. Chem. B 2004, 108, 1809–1811. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Kropman, M.F.; Bakker, H.J. Femtosecond mid-infrared spectroscopy of aqueous solvation shells. J. Chem. Phys. 2001, 115, 8942–8948. [Google Scholar] [CrossRef]
- Almora-Barrios, N.; de Leeuw, N.H. Modelling the interaction of a Hyp-Pro-Gly peptide with hydroxyapatite surfaces in aqueous environment. CrystEngComm 2010, 12, 960–967. [Google Scholar] [CrossRef]
- Ruiz-Hernandez, S.; Streeter, I.; de Leeuw, N.H. The effect of water on the binding of glycosaminoglycan saccharides to hydroxyapatite surfaces: A molecular dynamics study. Phys. Chem. Chem. Phys. 2015, 17, 22377–22388. [Google Scholar] [CrossRef] [PubMed]
- Rabone, J.A.L.; de Leeuw, N.H. Interatomic potential models for natural apatite crystals: Incorporating strontium and the lanthanides. J. Comput. Chem. 2006, 27, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.X. Mechanism and Thermodynamics of Ion Selectivity in Aqueous Solutions of 18-Crown-6 Ether: A Molecular Dynamics Study. J. Am. Chem. Soc. 1995, 117, 6954–6960. [Google Scholar] [CrossRef]
- Chandra, A. Dynamical Behavior of Anion−Water and Water−Water Hydrogen Bonds in Aqueous Electrolyte Solutions: A Molecular Dynamics Study. J. Phys. Chem. B 2003, 107, 3899–3906. [Google Scholar] [CrossRef]
- Fennell, C.J.; Bizjak, A.; Vlachy, V.; Dill, K.A.; Sarupria, S.; Rajamani, S.; Garde, S. Ion Pairing in Molecular Simulations of Aqueous Alkali Halide Solutions. J. Phys. Chem. B 2009, 113, 14837–14838. [Google Scholar] [CrossRef]
- Gujt, J.; Bešter-Rogač, M.; Hribar-Lee, B. An Investigation of Ion-Pairing of Alkali Metal Halides in Aqueous Solutions Using the Electrical Conductivity and the Monte Carlo Computer Simulation Methods. J. Mol. Liq. 2014, 190, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Rasaiah, J.C. Molecular dynamics simulation of ion mobility. 2. Alkali metal and halide ions using the SPC/E model for water at 25 °C. J. Phys. Chem. 1996, 100, 1420–1425. [Google Scholar] [CrossRef]
- Nahtigal, I.G.; Zasetsky, A.Y.; Svishchev, I.M. Nucleation of NaCl Nanoparticles in Supercritical Water: Molecular Dynamics Simulations. J. Phys. Chem. B 2008, 112, 7537–7543. [Google Scholar] [CrossRef] [PubMed]
- Sudarsanan, K.T.; Young, R. Significant precision in crystal structure details. Holly Springs hydroxyapatite. Acta Crystallogr. Sect. B 1969, 25, 1534–1543. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulations of Liquids; Oxford Science Publications: Oxford, UK, 1987. [Google Scholar]
- Chandra, A. Effects of Ion Atmosphere on Hydrogen-Bond Dynamics in Aqueous Electrolyte Solutions. Phys. Rev. Lett. 2000, 85, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.S.; Tran, H.T.; Schwenk, C.F.; Rode, B.M. Characterization of dynamics and reactivities of solvated ions by ab initio simulations. J. Comput. Chem. 2004, 25, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, D.; de Leeuw, N.H. First principles simulations of the structural and dynamical properties of hydrated metal ions Me2+ and solvated metal carbonates (Me = Ca, Mg and Sr). Cryst. Growth Des. 2010, 10, 4292–4302. [Google Scholar] [CrossRef]
- Wolthers, M.; Di Tommaso, D.; Du, Z.; de Leeuw, N.H. Variations in calcite growth kinetics with surface topography: Molecular dynamics simulations and process-based growth kinetics modelling. CrystEngComm 2013, 15, 5506–5514. [Google Scholar] [CrossRef]
- Soper, A.K.; Bruni, F.; Ricci, M.A. Site-site pair correlation functions of water from 25 to 400 °C: Revised analysis of new and old diffraction data. J. Chem. Phys. 1997, 106, 247–254. [Google Scholar] [CrossRef]
- Mahoney, M.W.; Jorgensen, W.L. Diffusion constant of the TIP5P model liquid water. J. Chem. Phys. 2001, 114, 363–366. [Google Scholar] [CrossRef]
- Di Tommaso, D.; Ruiz-Agudo, E.; de Leeuw, N.H.; Putnis, A.; Putnis, C.V. Modelling the effects of salt solutions on the hydration of calcium ions. Phys. Chem. Chem. Phys. 2014, 16, 7772–7785. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, P.; Gale, J.D.; Quigley, D.; Rodger, P.M. Derivation of an accurate force-field for simulating the growth of calcium carbonate from aqueous solution: A new model for the calcite—Water interface. J. Phys. Chem. C 2010, 114, 5997–6010. [Google Scholar] [CrossRef]
- Hirunsit, P.; Balbuena, P.B. The effects of confinement on water structure and dynamics: A molecular simulation study. J. Phys. Chem. C 2007, 111, 1709–1715. [Google Scholar] [CrossRef]
- Mosaddeghi, H.; Alavi, S.; Kowsari, M.H.; Najafi, B. Simulations of structural and dynamic anisotropy in nano-confined water between parallel graphite plates. J. Chem. Phys. 2012, 137, 184703. [Google Scholar] [CrossRef] [PubMed]
- Boon, J.P.; Yip, S. Molecular Hydrodynamics; McGraw-Hill International Book Co.: New York, NY, USA, 1980. [Google Scholar]
- Demontis, P.; Suffritti, G.B.; Tilocca, A. Diffusion and vibrational relaxation of a diatomic molecule in the pore network of a pure silica zeolite: A molecular dynamics study. J. Chem. Phys. 1996, 105, 5586. [Google Scholar] [CrossRef]
- Tian, K.V.; Chass, G.; Di Tommaso, D. Simulations reveal the role of composition into the atomic-level flexibility of bioactive glass cements. Phys. Chem. Chem. Phys. 2016, 18, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Stillinger, F.H.; Raman, A. Improved Simulation of Liquid Water by Molecular Dynamics. J. Chem. Phys. 1974, 60, 1545. [Google Scholar] [CrossRef]
- Ockwig, N.W.; Cygan, R.T.; Criscenti, L.J.; Nenoff, T.M. Incoherent inelastic neutron scattering studies of nanoconfined water in clinoptilolite and heulandite zeolites. J. Phys. Chem. C. 2008, 112, 13629–13634. [Google Scholar] [CrossRef]
- Corsaro, C.; Crupi, V.; Majolino, D.; Parker, S.F.; Venuti, V.; Wanderlinght, U. Inelastic Neutron Scattering Study of Water in Hydrated LTA-Type Zeolites. J. Phys. Chem. A. 2006, 110, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Tayal, V.P.; Srivastava, B.K.; Khandelwal, D.P. Librational Modes of Crystal Water in Hydrated Solids. Appl. Spectrosc. Rev. 1980, 16, 43–134. [Google Scholar] [CrossRef]
- Schwenk, C.F.; Hofer, T.S.; Randolf, B.R.; Rode, B.M. The influence of heteroligands on the reactivity of Ni2+ in solution. Phys. Chem. Chem. Phys. 2005, 7, 1669–1673. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
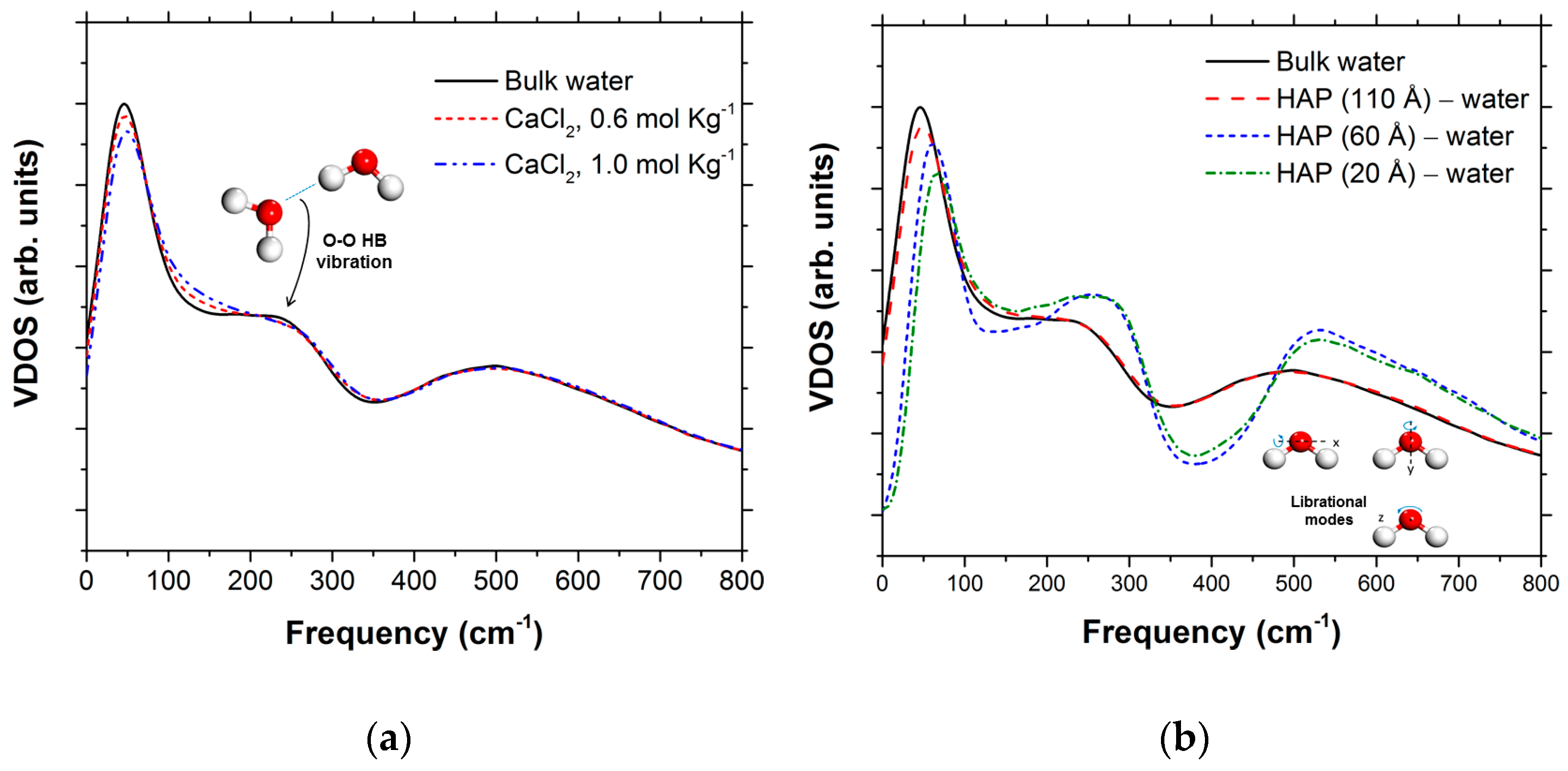{kind=link}
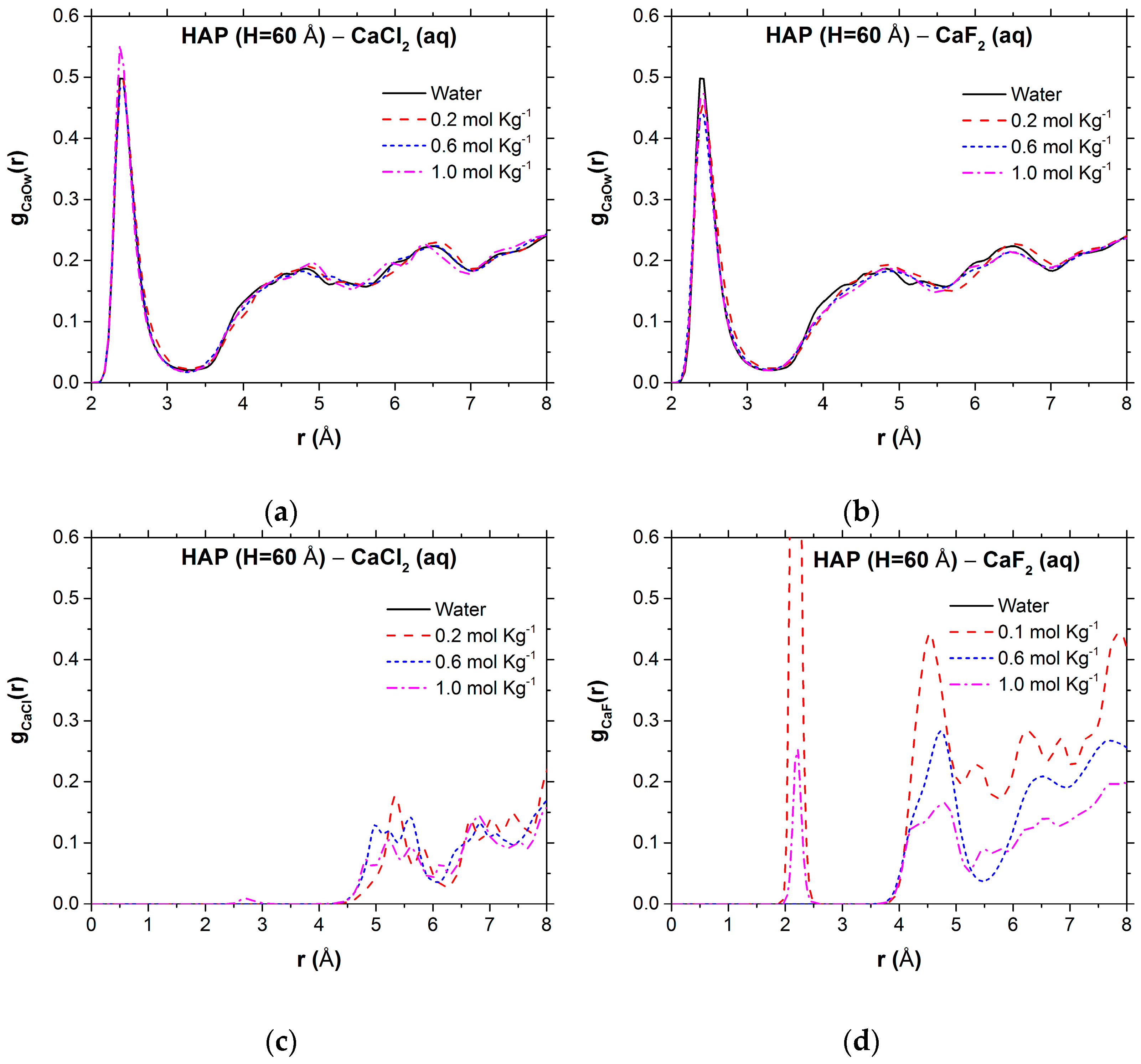{kind=link}
| Number of Ca10(PO4)6(OH)2 Units | Number of CaX2 | Number of H2O | b | |
|---|---|---|---|---|
| Solution | ||||
| Water | – | – | 729 | 0.0 |
| CaCl2 | – | 8 | 705 | 0.6 |
| – | 16 | 681 | 1.3 | |
| CaF2 | – | 8 | 705 | 0.6 |
| – | 16 | 681 | 1.3 | |
| HAP, H = 110 Å | ||||
| Water | 36 | – | 2505 | 0.0 |
| HAP, H = 60 Å | ||||
| Water | 36 | – | 1363 | 0.0 |
| CaCl2 | 36 | 4 | 1351 | 0.2 |
| 36 | 11 | 1330 | 0.5 | |
| 36 | 23 | 1294 | 1.0 | |
| CaF2 | 36 | 4 | 1351 | 0.2 |
| 36 | 11 | 1330 | 0.5 | |
| 36 | 23 | 1294 | 1.0 | |
| HAP, H = 40 Å | ||||
| Water | 36 | – | 910 | 0.0 |
| HAP, H = 20 Å | ||||
| Water | 36 | – | 455 | 0.0 |
| Number of Hydrogen Bonds (%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| b | 0 | 1 | 2 | 3 | 4 | 5 | Average | |
| Bulk solution | ||||||||
| Water | – | 0.0 | 0.9 | 8.8 | 33.0 | 51.3 | 5.9 | 3.53 |
| CaCl2 | 0.6 | 0.2 | 2.7 | 14.5 | 34.0 | 43.7 | 4.8 | 3.30 |
| 1.3 | 0.4 | 4.7 | 19.2 | 34.0 | 37.6 | 4.0 | 3.16 | |
| CaF2 | 0.6 | 0.2 | 2.8 | 13.5 | 33.4 | 44.7 | 5.3 | 3.36 |
| 1.3 | 0.5 | 4.7 | 17.1 | 33.1 | 39.6 | 4.8 | 3.21 | |
| HAP, H = 110 Å | ||||||||
| Water | – | 0.4 | 2.8 | 11.1 | 32.8 | 47.4 | 5.5 | 3.41 |
| HAP, H = 60 Å | ||||||||
| Water | – | 0.5 | 3.9 | 8.4 | 20.6 | 61.9 | 4.7 | 3.54 |
| CaCl2 | 0.2 | 0.7 | 4.5 | 10.0 | 22.0 | 58.4 | 4.5 | 3.46 |
| 0.5 | 0.6 | 5.0 | 13.3 | 24.8 | 52.1 | 4.0 | 3.35 | |
| 1.0 | 0.9 | 6.8 | 16.8 | 26.9 | 45.2 | 3.4 | 3.19 | |
| CaF2 | 0.2 | 0.9 | 5.3 | 11.1 | 27.1 | 50.4 | 5.1 | 3.36 |
| 0.5 | 1.1 | 6.1 | 15.2 | 30.6 | 41.9 | 5.0 | 3.22 | |
| 1.0 | 1.7 | 8.6 | 17.2 | 27.3 | 40.8 | 4.2 | 3.10 | |
| HAP, H = 40 Å | ||||||||
| Water | – | 0.9 | 6.7 | 8.5 | 19.4 | 60.0 | 4.3 | 3.44 |
| HAP, H = 20 Å | ||||||||
| Water | – | 0.8 | 11.2 | 12.4 | 12.4 | 59.7 | 3.5 | 3.29 |
| b | D W × 10−10 (m2·s−1) | |
|---|---|---|
| Solution | ||
| Water | – | 25.8 |
| CaCl2 | 0.6 | 21.1 |
| 1.3 | 16.3 | |
| CaF2 | 0.6 | 22.3 |
| 1.3 | 17.1 | |
| HAP, H = 110 Å | ||
| Water | – | 21.9 |
| HAP, H = 60 Å | ||
| Water | – | 5.4 |
| CaCl2 | 0.2 | 3.9 |
| 0.5 | 3.5 | |
| 1.0 | 2.9 | |
| CaF2 | 0.2 | 6.6 |
| 0.5 | 5.3 | |
| 1.0 | 3.9 | |
| HAP, H = 40 Å | ||
| Water | – | 2.9 |
| HAP, H = 20 Å | ||
| Water | – | 0.2 |
| b | tsim (ps) | Nex | Nex/100 ps | log kw (s−1 ) a | |
|---|---|---|---|---|---|
| Solution | |||||
| Ca2+ | – | 8000 | 1532 | 19.2 | 11.3 |
| CaCl2 | 0.6 | 8000 | 396.8 | 5.0 | 10.7 |
| 1.3 | 8000 | 225.1 | 2.8 | 10.4 | |
| CaF2 | 0.6 | 8000 | 308.1 | 3.9 | 10.6 |
| 1.3 | 8000 | 202.5 | 2.5 | 10.4 | |
| HAP, H = 110 Å | |||||
| Water | – | 2000 | 317.3 | 15.9 | 11.2 |
| HAP, H = 60 Å b | |||||
| Water | – | 4000 | 337.6 | 8.4 | 10.9 |
| CaCl2 | 0.2 | 4000 | 374.4 | 9.4 | 11.0 |
| 0.5 | 4000 | 312.4 | 7.8 | 10.9 | |
| 1.0 | 4000 | 248.6 | 6.2 | 10.8 | |
| CaF2 | 0.2 | 3000 | 336.9 | 11.2 | 11.1 |
| 0.5 | 3000 | 296.7 | 9.9 | 11.0 | |
| 1.0 | 3000 | 278.4 | 9.3 | 11.0 | |
| HAP, H = 40 Å | |||||
| Water | – | 4000 | 367.4 | 9.2 | 11.0 |
| HAP, H = 20 Å | |||||
| Water | – | 4000 | 111.9 | 2.8 | 10.4 |
| HAP, H = 60 Å | b | Ca-X Surface Coverage (%) | Speciation of MX Pairs | ||||
|---|---|---|---|---|---|---|---|
| CIP | SSHIP | SSIP | FIP | ||||
| CaCl2 | 0.2 | 1.9 | 0.0 | 11.5 | 21.5 | 24.4 | 42.6 |
| 0.5 | 1.8 | 0.0 | 18.9 | 29.4 | 36.7 | 15.0 | |
| 1.0 | 1.7 | 0.1 | 63.2 | 19.9 | 15.4 | 1.5 | |
| CaF2 | 0.2 | 2.0 | 1.5 | 21.6 | 27.6 | 23.2 | 27.6 |
| 0.5 | 1.8 | 0.0 | 28.9 | 48.7 | 19.7 | 2.8 | |
| 1.0 | 1.8 | 1.3 | 28.6 | 60.1 | 10.2 | 1.1 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Tommaso, D.; Prakash, M.; Lemaire, T.; Lewerenz, M.; De Leeuw, N.H.; Naili, S. Molecular Dynamics Simulations of Hydroxyapatite Nanopores in Contact with Electrolyte Solutions: The Effect of Nanoconfinement and Solvated Ions on the Surface Reactivity and the Structural, Dynamical, and Vibrational Properties of Water. Crystals 2017, 7, 57. https://doi.org/10.3390/cryst7020057
Di Tommaso D, Prakash M, Lemaire T, Lewerenz M, De Leeuw NH, Naili S. Molecular Dynamics Simulations of Hydroxyapatite Nanopores in Contact with Electrolyte Solutions: The Effect of Nanoconfinement and Solvated Ions on the Surface Reactivity and the Structural, Dynamical, and Vibrational Properties of Water. Crystals. 2017; 7(2):57. https://doi.org/10.3390/cryst7020057
Chicago/Turabian StyleDi Tommaso, Devis, Muthuramalingam Prakash, Thibault Lemaire, Marius Lewerenz, Nora H. De Leeuw, and Salah Naili. 2017. "Molecular Dynamics Simulations of Hydroxyapatite Nanopores in Contact with Electrolyte Solutions: The Effect of Nanoconfinement and Solvated Ions on the Surface Reactivity and the Structural, Dynamical, and Vibrational Properties of Water" Crystals 7, no. 2: 57. https://doi.org/10.3390/cryst7020057
APA StyleDi Tommaso, D., Prakash, M., Lemaire, T., Lewerenz, M., De Leeuw, N. H., & Naili, S. (2017). Molecular Dynamics Simulations of Hydroxyapatite Nanopores in Contact with Electrolyte Solutions: The Effect of Nanoconfinement and Solvated Ions on the Surface Reactivity and the Structural, Dynamical, and Vibrational Properties of Water. Crystals, 7(2), 57. https://doi.org/10.3390/cryst7020057