
Two-Stage Crystallizer Design for High Loading of Poorly Water-Soluble Pharmaceuticals in Porous Silica Matrices


Abstract
:1. Introduction
2. Results
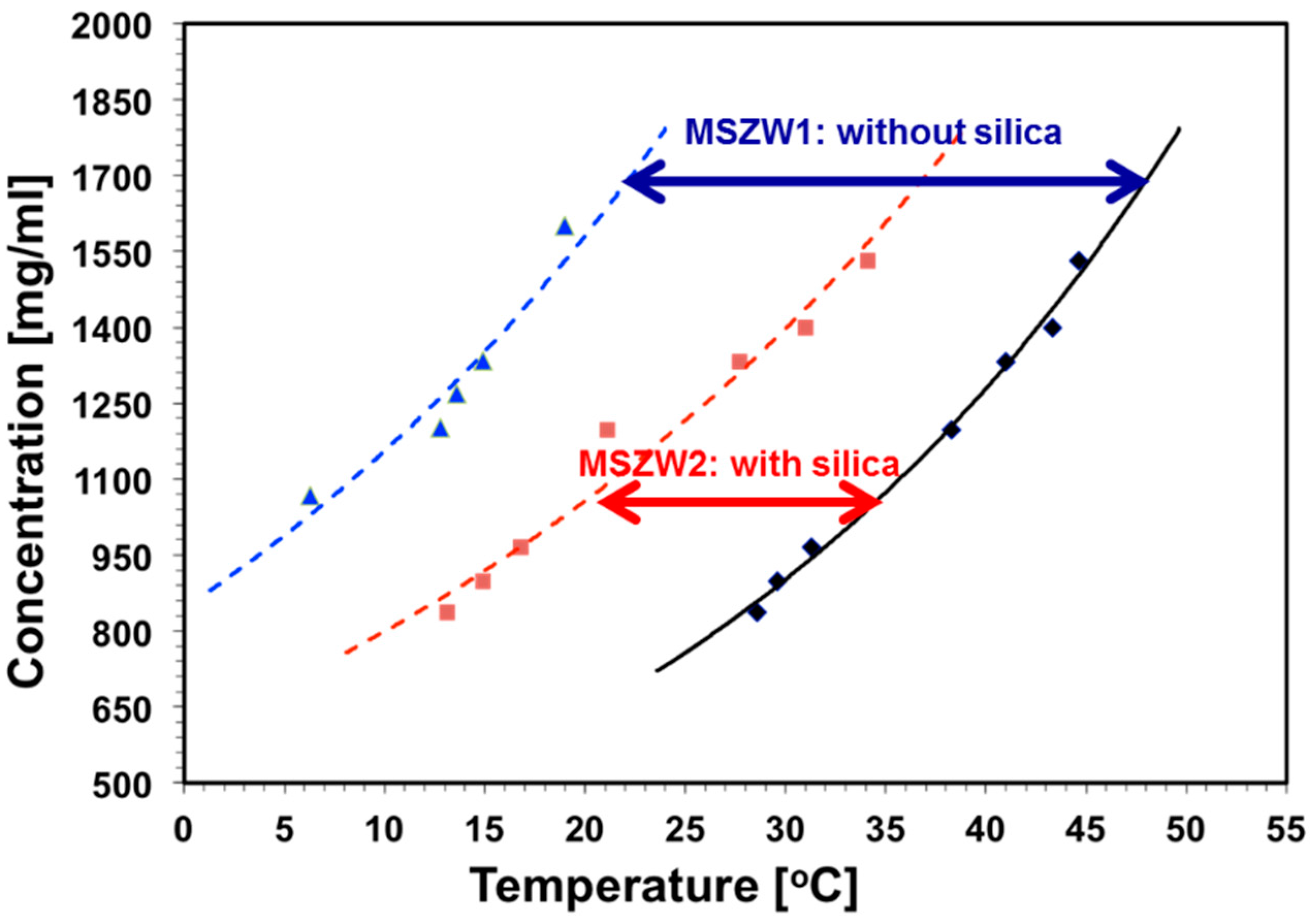
2.1. Selection of MSMPR Operating Parameters
2.2. Analysis of FEN Loading in MSMPR Experiments
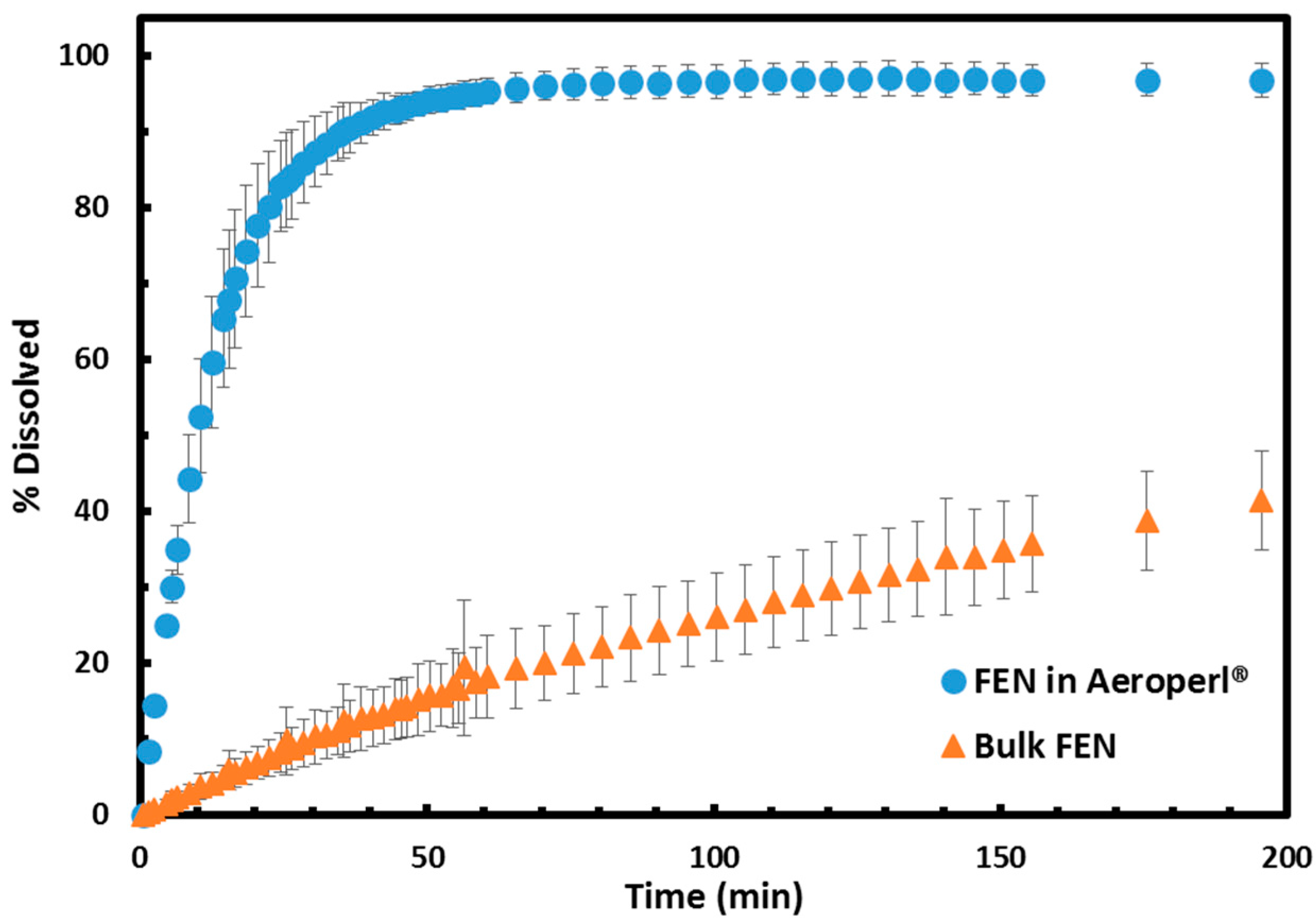
2.3. Dissolution Profile Enhancement
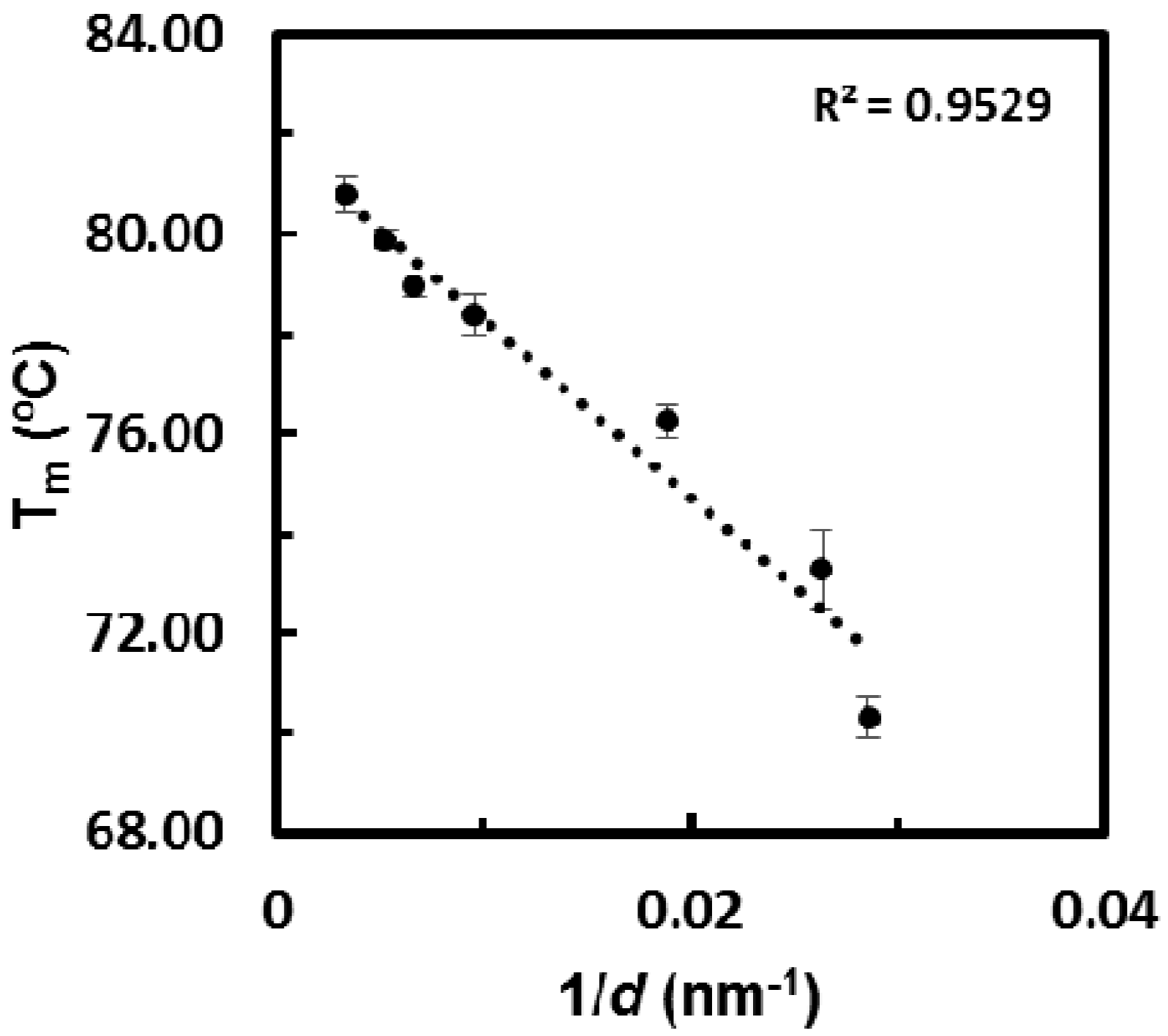
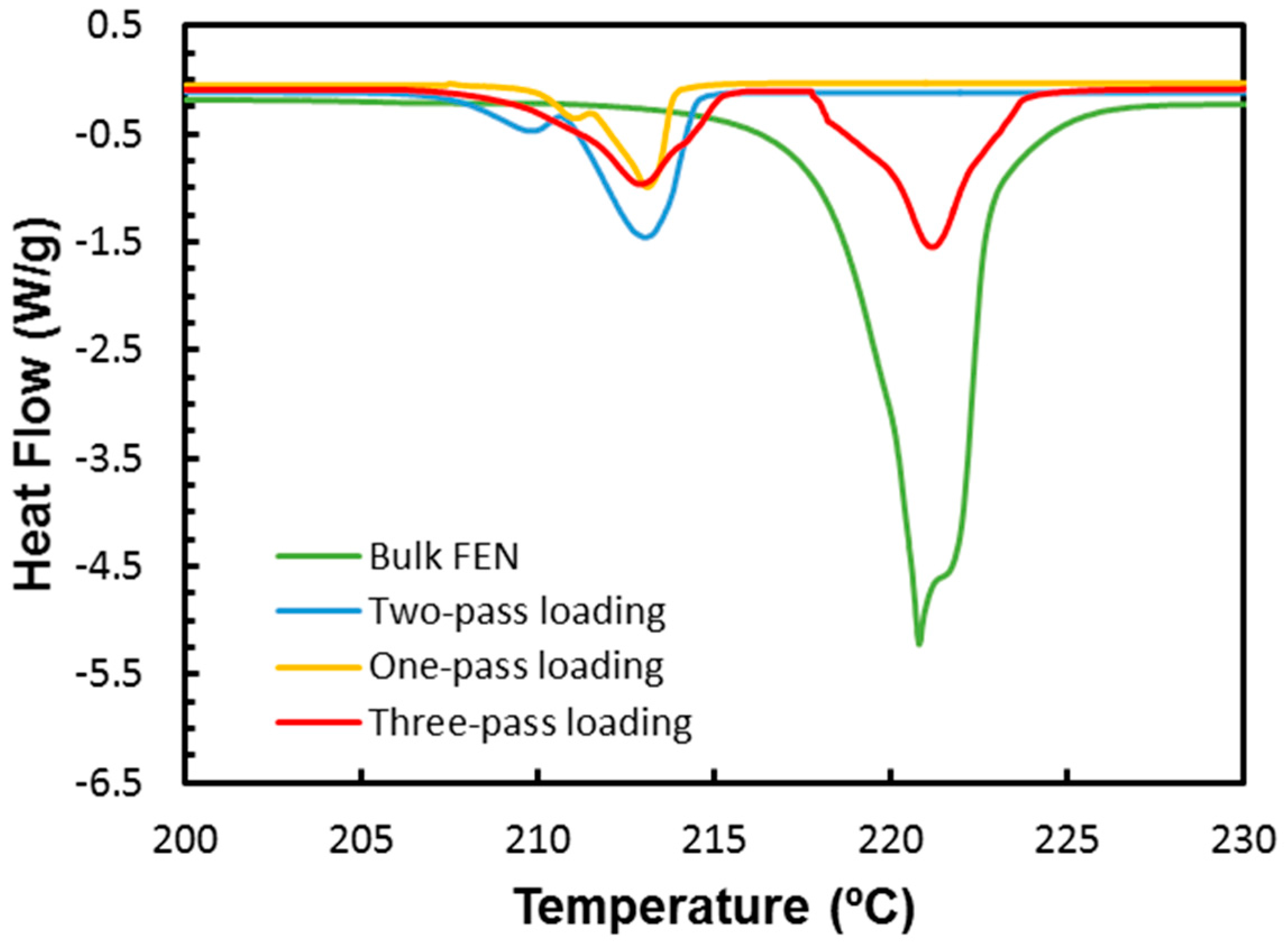
2.4. Melting Point Depression Analysis of Nanocrystals with DSC
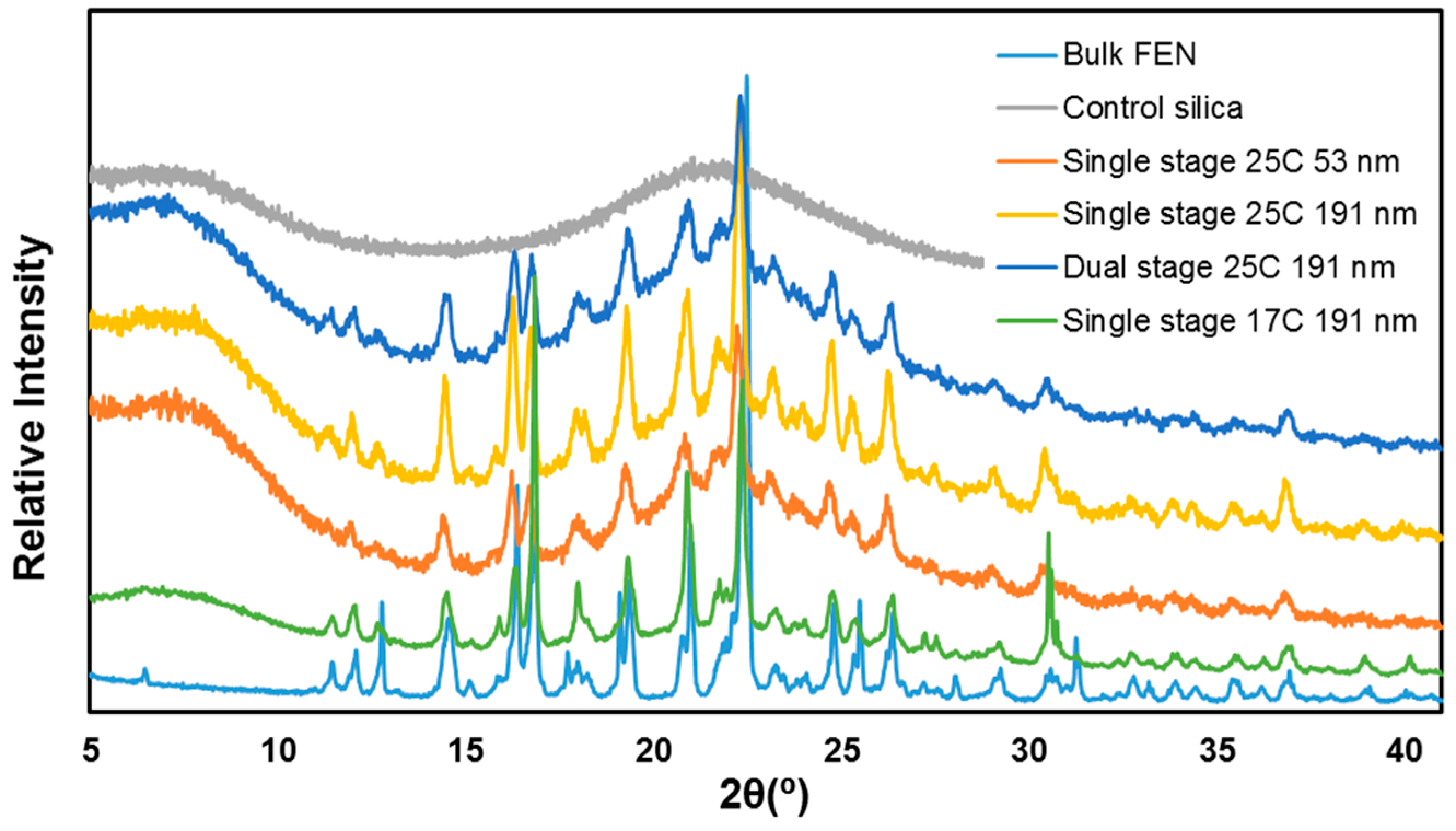
2.5. Crystal Form Identification with X-ray Powder Diffraction (XRPD)
2.6. Extension of Principle to Poorly Soluble Compounds
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Experimental Apparatus
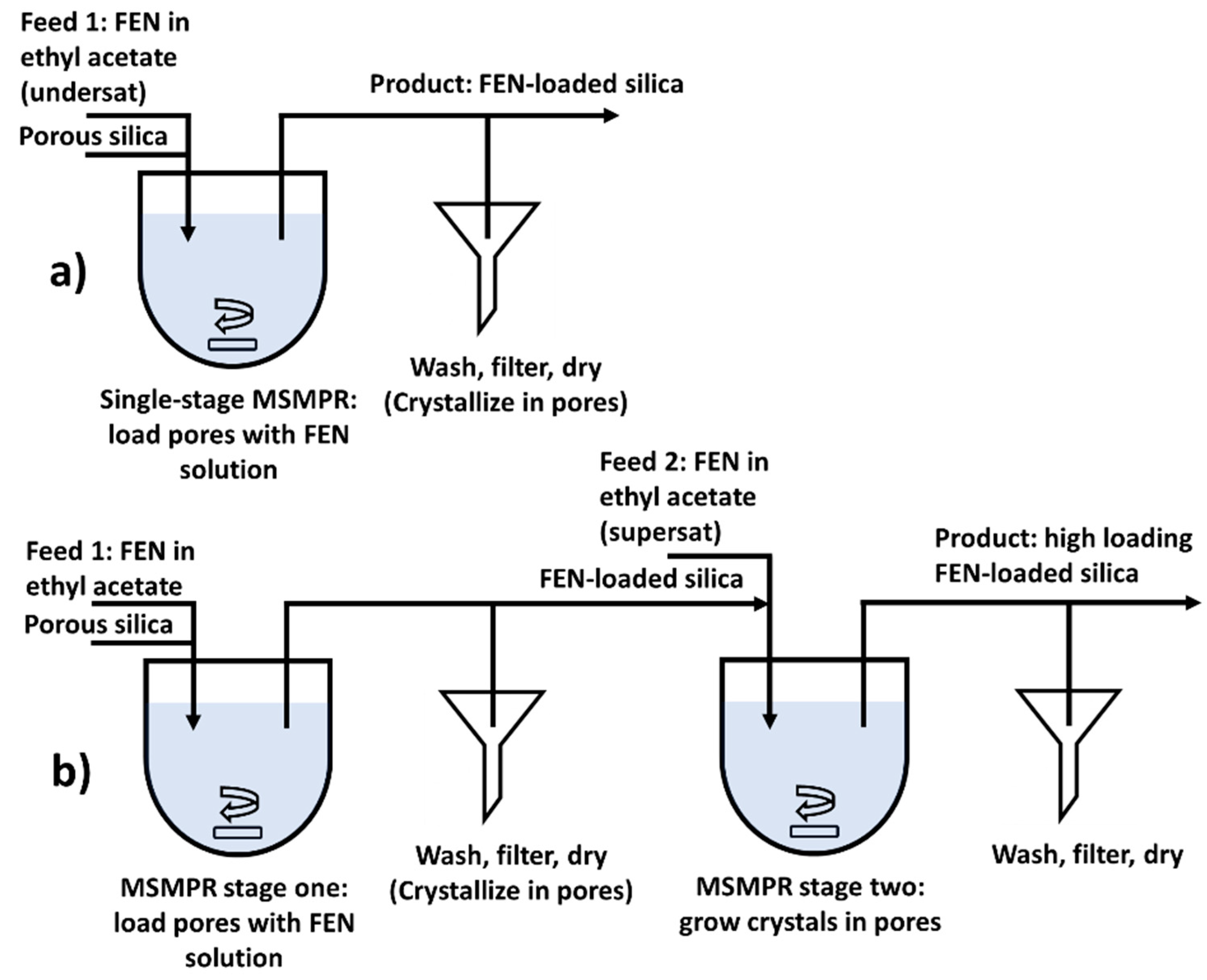
4.2.1. Single-Stage MSMPR
4.2.2. Two-Stage MSMPR
4.2.3. Extension of Principle to Poorly Water-Soluble Compounds
4.3. Analytical Techniques
4.3.1. X-ray Powder Diffraction
4.3.2. Thermogravimetric Analysis
4.3.3. Differential Scanning Calorimetry
4.3.4. Solubility Measurements
4.3.5. Dissolution Testing
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stegemann, S.; Leveiller, F.; Franchi, D.; de Jong, H.; Lindén, H. When poor solubility becomes an issue: From early stage to proof of concept. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2007, 31, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Otto, D.P.; de Villiers, M.M. Physicochemical Principles of Nanosized Drug Delivery Systems. In Nanotechnology in Drug Delivery; de Villiers, M.M., Aramwit, P., Kwon, G.S., Eds.; Biotechnology: Pharmaceutical Aspects; Springer: New York, NY, USA, 2009; pp. 3–33. [Google Scholar]
- Shegokar, R.; Müller, R.H. Nanocrystals: Industrially feasible multifunctional formulation technology for poorly soluble actives. Int. J. Pharm. 2010, 399, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Peters, K. Nanosuspensions for the formulation of poorly soluble drugs: I. Preparation by a size-reduction technique. Int. J. Pharm. 1998, 160, 229–237. [Google Scholar] [CrossRef]
- Zuo, B.; Sun, Y.; Li, H.; Liu, X.; Zhai, Y.; Sun, J.; He, Z. Preparation and in vitro/in vivo evaluation of fenofibrate nanocrystals. Int. J. Pharm. 2013, 455, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Mullin, J.W. 3—Solutions and solubility. In Crystallization, 4th ed.; Butterworth-Heinemann: Oxford, UK, 2001; pp. 86–134. [Google Scholar]
- Kim, K.; Lee, I.S.; Centrone, A.; Hatton, T.A.; Myerson, A.S. Formation of Nanosized Organic Molecular Crystals on Engineered Surfaces. J. Am. Chem. Soc. 2009, 131, 18212–18213. [Google Scholar] [CrossRef] [PubMed]
- Junghanns, J.-U.A.H.; Müller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–310. [Google Scholar]
- Hu, J.; Johnston, K.P.; Williams, R.O., 3rd. Nanoparticle Engineering Processes for Enhancing the Dissolution Rates of Poorly Water Soluble Drugs. Drug Dev. Ind. Pharm. 2004, 30, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Keck, C.M.; Müller, R.H. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur. J. Pharm. Biopharm. 2006, 62, 3–16. [Google Scholar] [CrossRef] [PubMed]
- List, M.; Sucker, H. Hydrosols of Pharmacologically Active Agents and Their Pharmaceutical Compositions Comprising Them. U.S. Patent 5,389,382 A, 14 February 1995. [Google Scholar]
- De Waard, H.; Grasmeijer, N.; Hinrichs, W.L.J.; De Beer, T.; Frijlink, H.W. A Process Suitable for Large-Scale Production of Drug Nanocrystals. Pharm. Technol. 2011, 35, 58–62. [Google Scholar]
- Lee, S.; Nam, K.; Kim, M.S.; Jun, S.W.; Park, J.-S.; Woo, J.S.; Hwang, S.-J. Preparation and characterization of solid dispersions of itraconazole by using aerosol solvent extraction system for improvement in drug solubility and bioavailability. Arch. Pharm. Res. 2005, 28, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.L.; Johnston, K.P.; Williams, R.O., 3rd. Solution-Based Particle Formation of Pharmaceutical Powders by Supercritical or Compressed Fluid Co2 and Cryogenic Spray-Freezing Technologies. Drug Dev. Ind. Pharm. 2001, 27, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Sarkari, M.; Brown, J.; Chen, X.; Swinnea, S.; Williams, R.O., III; Johnston, K.P. Enhanced drug dissolution using evaporative precipitation into aqueous solution. Int. J. Pharm. 2002, 243, 17–31. [Google Scholar] [CrossRef]
- Chen, X.; Young, T.J.; Sarkari, M.; Williams, R.O., III; Johnston, K.P. Preparation of cyclosporine A nanoparticles by evaporative precipitation into aqueous solution. Int. J. Pharm. 2002, 242, 3–14. [Google Scholar] [CrossRef]
- Panagiotou, T.; Mesite, S.V.; Fisher, R.J. Production of Norfloxacin Nanosuspensions Using Microfluidics Reaction Technology through Solvent/Antisolvent Crystallization. Ind. Eng. Chem. Res. 2009, 48, 1761–1771. [Google Scholar] [CrossRef]
- Chan, H.-K.; Kwok, P.C.L. Production methods for nanodrug particles using the bottom-up approach. Adv. Drug Deliv. Rev. 2011, 63, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; de Matas, M.; Zhang, J.; Anwar, J. Nanocrystal Preparation: Low-Energy Precipitation Method Revisited. Cryst. Growth Des. 2013, 13, 2766–2777. [Google Scholar] [CrossRef]
- Möschwitzer, J.P. Drug nanocrystals in the commercial pharmaceutical development process. Int. J. Pharm. 2013, 453, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.J.K.; Slipper, I.; Walunj, A.; Jain, A.; Favretto, M.E.; Kallinteri, P.; Douroumis, D. Inclusion of poorly soluble drugs in highly ordered mesoporous silica nanoparticles. Int. J. Pharm. 2010, 387, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.K.; Bogner, R.H. Application of Mesoporous Silicon Dioxide and Silicate in Oral Amorphous Drug Delivery Systems. J. Pharm. Sci. 2012, 101, 444–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, S. Ordered mesoporous materials for drug delivery. Microporous Mesoporous Mater. 2009, 117, 1–9. [Google Scholar] [CrossRef]
- Azaïs, T.; Tourné-Péteilh, C.; Aussenac, F.; Baccile, N.; Coelho, C.; Devoisselle, J.-M.; Babonneau, F. Solid-State NMR Study of Ibuprofen Confined in MCM-41 Material. Chem. Mater. 2006, 18, 6382–6390. [Google Scholar] [CrossRef]
- Azad, M.; Moreno, J.; Bilgili, E.; Davé, R. Fast dissolution of poorly water soluble drugs from fluidized bed coated nanocomposites: Impact of carrier size. Int. J. Pharm. 2016, 513, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Geszke-Moritz, M.; Moritz, M. APTES-modified mesoporous silicas as the carriers for poorly water-soluble drug. Modeling of diflunisal adsorption and release. Appl. Surf. Sci. 2016, 368, 348–359. [Google Scholar] [CrossRef]
- Martín, A.; García, R.A.; Karaman, D.S.; Rosenholm, J.M. Polyethyleneimine-functionalized large pore ordered silica materials for poorly water-soluble drug delivery. J. Mater. Sci. 2014, 49, 1437–1447. [Google Scholar] [CrossRef]
- Dwyer, L.; Michaelis, V.; O’Mahony, M.; Griffin, R.; Myerson, A. Confined crystallization of fenofibrate in nanoporous silica. CrystEngComm 2015, 17, 7922–7929. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.L.; McKenna, G.B. Vitrification and Crystallization of Organic Liquids Confined to Nanoscale Pores. Chem. Mater. 1996, 8, 2128–2137. [Google Scholar] [CrossRef]
- Ha, J.-M.; Hamilton, B.D.; Hillmyer, M.A.; Ward, M.D. Phase Behavior and Polymorphism of Organic Crystals Confined within Nanoscale Chambers. Cryst. Growth Des. 2009, 9, 4766–4777. [Google Scholar] [CrossRef]
- Ha, J.-M.; Wolf, J.H.; Hillmyer, M.A.; Ward, M.D. Polymorph Selectivity under Nanoscopic Confinement. J. Am. Chem. Soc. 2004, 126, 3382–3383. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberger, N.; Anders, N.; Golitsyn, Y.; Steinhart, M.; Enke, D.; Saalwächter, K.; Beiner, M. Pharmaceutical nanocrystals confined in porous host systems—Interfacial effects and amorphous interphases. Chem. Commun. 2016, 52, 4466–4469. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ong, T.C.; Michaelis, V.K.; Heng, S.; Huang, J.; Griffin, R.G.; Myerson, A.S. Formation of organic molecular nanocrystals under rigid confinement with analysis by solid state NMR. CrystEngComm 2014, 16, 9345–9352. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Ward, M.D. Crystallization under nanoscale confinement. Chem. Soc. Rev. 2014, 43, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, G.T.; Enke, D.; Steinhart, M.; Beiner, M. Size-dependent growth of polymorphs in nanopores and Ostwald’s step rule of stages. Phys. Chem. Chem. Phys. 2011, 13, 21367–21374. [Google Scholar] [CrossRef] [PubMed]
- Graubner, G.; Rengarajan, G.T.; Anders, N.; Sonnenberger, N.; Enke, D.; Beiner, M.; Steinhart, M. Morphology of Porous Hosts Directs Preferred Polymorph Formation and Influences Kinetics of Solid/Solid Transitions of Confined Pharmaceuticals. Cryst. Growth Des. 2014, 14, 78–86. [Google Scholar] [CrossRef]
- Beiner, M.; Rengarajan; Pankaj, S.; Enke, D.; Steinhart, M. Manipulating the Crystalline State of Pharmaceuticals by Nanoconfinement. Nano Lett. 2007, 7, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Ahern, R.J.; Hanrahan, J.P.; Tobin, J.M.; Ryan, K.B.; Crean, A.M. Comparison of fenofibrate-mesoporous silica drug-loading processes for enhanced drug delivery. Eur. J. Pharm. Sci. 2013, 50, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, M.M.; Motawaa, A.M.; Samaha, M.W. Tadalafil inclusion in microporous silica as effective dissolution enhancer: Optimization of loading procedure and molecular state characterization. J. Pharm. Sci. 2011, 100, 1805–1818. [Google Scholar] [CrossRef] [PubMed]
- Van Speybroeck, M.; Barillaro, V.; Thi, T.D.; Mellaerts, R.; Martens, J.; Van Humbeeck, J.; Vermant, J.; Annaert, P.; Van den Mooter, G.; Augustijns, P. Ordered mesoporous silica material SBA-15: A broad-spectrum formulation platform for poorly soluble drugs. J. Pharm. Sci. 2009, 98, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Hillerström, A.; van Stam, J.; Andersson, M. Ibuprofen loading into mesostructured silica using liquid carbon dioxide as a solvent. Green Chem. 2009, 11, 662–667. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhi, Z.; Jiang, T.; Zhang, J.; Wang, Z.; Wang, S. Spherical mesoporous silica nanoparticles for loading and release of the poorly water-soluble drug telmisartan. J. Control. Release 2010, 145, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Junyaprasert, V.B.; Morakul, B. Nanocrystals for enhancement of oral bioavailability of poorly water-soluble drugs. Asian J. Pharm. Sci. 2015, 10, 13–23. [Google Scholar] [CrossRef]
- O’Mahony, M.; Leung, A.K.; Ferguson, S.; Trout, B.L.; Myerson, A.S. A Process for the Formation of Nanocrystals of Active Pharmaceutical Ingredients with Poor Aqueous Solubility in a Nanoporous Substrate. Org. Process. Res. Dev. 2015, 19, 1109–1118. [Google Scholar] [CrossRef]
- Alvarez, A.J.; Singh, A.; Myerson, A.S. Crystallization of Cyclosporine in a Multistage Continuous MSMPR Crystallizer. Cryst. Growth Des. 2011, 11, 4392–4400. [Google Scholar] [CrossRef]
- Peña, R.; Nagy, Z.K. Process Intensification through Continuous Spherical Crystallization Using a Two-Stage Mixed Suspension Mixed Product Removal (MSMPR) System. Cryst. Growth Des. 2015, 15, 4225–4236. [Google Scholar] [CrossRef]
- Li, J.; Trout, B.L.; Myerson, A.S. Multistage Continuous Mixed-Suspension, Mixed-Product Removal (MSMPR) Crystallization with Solids Recycle. Org. Process. Res. Dev. 2016, 20, 510–516. [Google Scholar] [CrossRef]
- Lai, T.-T.C.; Cornevin, J.; Ferguson, S.; Li, N.; Trout, B.L.; Myerson, A.S. Control of Polymorphism in Continuous Crystallization via Mixed Suspension Mixed Product Removal Systems Cascade Design. Cryst. Growth Des. 2015, 15, 3374–3382. [Google Scholar] [CrossRef]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Heinz, A.; Gordon, K.C.; McGoverin, C.M.; Rades, T.; Strachan, C.J. Understanding the solid-state forms of fenofibrate--a spectroscopic and computational study. Eur. J. Pharm. Biopharm. 2009, 71, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Shen, S.; Tan, D.C.T.; Ng, W.K.; Liu, X.; Chia, L.S.O.; Irwan, A.W.; Tan, R.; Nowak, S.A.; Marsh, K.; et al. High drug load, stable, manufacturable and bioavailable fenofibrate formulations in mesoporous silica: A comparison of spray drying versus solvent impregnation methods. Drug Deliv. 2016, 23, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.-M.; Hillmyer, M.A.; Ward, M.D. Thermotropic properties of organic nanocrystals embedded in ultrasmall crystallization chambers. J. Phys. Chem. B 2005, 109, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- O′Neil, M. The Merck Index—An Encyclopedia of Chemicals, Drugs, and Biologicals; Royal Society of Chemistry: Cambridge, UK, 2013. [Google Scholar]
- Hu, G.; Li, H.; Wang, X.; Zhang, Y. Measurement and Correlation of Griseofulvin Solubility in Different Solvents at Temperatures from (281.95 to 357.60) K. J. Chem. Eng. Data 2010, 55, 3969–3971. [Google Scholar] [CrossRef]
- Elworthy, P.H.; Lipscomb, F.J. The effect of some non-ionic surfactants and a polyoxyethylene glycol on the dissolution rate of griseofulvin. J. Pharm. Pharmacol. 1968, 20, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Limnell, T.; Santos, H.A.; Mäkilä, E.; Heikkilä, T.; Salonen, J.; Murzin, D.Y.; Kumar, N.; Laaksonen, T.; Peltonen, L.; Hirvonen, J. Drug delivery formulations of ordered and nonordered mesoporous silica: Comparison of three drug loading methods. J. Pharm. Sci. 2011, 100, 3294–3306. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pore Size (nm) | Specific Surface Area (m2/g) | Pore Vol. (mL/g) | Theoretical Single-Stage Max. Loading from FEN Soln. (wt %) | Single-Stage Loading (wt %) | Theoretical Filled Pore Max. FEN Loading (wt %) | Two-Stage Loading (wt %) |
|---|---|---|---|---|---|---|
| 300 | 10 | 1.0 | 37.5 | 31.4 ± 1.7 | 54.1 | 41.6 ± 1.0 |
| 191 | 30 | 1.5 | 47.4 | 36 ± 1.9 | 63.9 | 50.2 ± 1.8 |
| 151 | 31 | 1.2 | 41.9 | 36.1 ± 1.7 | 58.6 | 40.2 ± 2.5 |
| 105 | 52 | 1.4 | 45.7 | 36.5 ± 4.2 | 62.3 | 54.8 ± 3.7 |
| 53 | 94 | 1.3 | 43.8 | 39.1 ± 1.0 | 60.5 | 55.6 ± 2.2 |
| 38 | 138 | 1.3 | 43.8 | 40.3 ± 2.3 | 60.5 | 56.1 ± 3.5 |
| 35 | 300 | 1.6 | 49.0 | - | 65.4 | 56.7 ± 1.6 |
| Temp. (°C) | Loading (wt %) | Notes from DSC Thermogram | |||
|---|---|---|---|---|---|
| Trial 1 | Trial 2 | Trial 3 | Avg. | ||
| 25 | 36.0 | 42.3 | 37.2 | 38.5 ± 3.5 | Single peak, confined crystals |
| 20 | 27.4 | 30.3 | 31.8 | 29.8 ± 2.2 | Single peak, confined crystals |
| 18 | 50.8 | 48.7 | 51.6 | 50.4 ± 1.5 | Single peak, confined crystals |
| 17 | 61.0 | 58.9 | 56.9 | 58.9 ± 2.1 | Single peak, confined crystals |
| 15 | 64.6 | 61.0 | 65.7 | 63.8 ± 2.5 | Two peaks, confined and surface crystals |
| API | Theoret. One-Pass Max. Load from 30 mg/mL Soln. (wt %) | Theoret. Filled Pore Max. Load (wt %) | One-Pass Loading (wt %) | Two-Pass Loading (wt %) | Three-Pass Loading (wt %) |
|---|---|---|---|---|---|
| GSF | 4.6 | 69.1 | 3.0 ± 0.6 | 16.9 ± 1.7 | 26.1 ± 2.0 |
| DSC notes | No surface crystals | No surface crystals | Some surface crystals | ||
| IMC | 4.6 | 61.5 | - | 10.9 ± 2.2 | 13.2 ± 3.9 |
| DSC notes | Undetected | Mostly surface crystals | Mostly surface crystals |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dwyer, L.; Kulkarni, S.; Ruelas, L.; Myerson, A. Two-Stage Crystallizer Design for High Loading of Poorly Water-Soluble Pharmaceuticals in Porous Silica Matrices. Crystals 2017, 7, 131. https://doi.org/10.3390/cryst7050131
Dwyer L, Kulkarni S, Ruelas L, Myerson A. Two-Stage Crystallizer Design for High Loading of Poorly Water-Soluble Pharmaceuticals in Porous Silica Matrices. Crystals. 2017; 7(5):131. https://doi.org/10.3390/cryst7050131
Chicago/Turabian StyleDwyer, Leia, Samir Kulkarni, Luzdary Ruelas, and Allan Myerson. 2017. "Two-Stage Crystallizer Design for High Loading of Poorly Water-Soluble Pharmaceuticals in Porous Silica Matrices" Crystals 7, no. 5: 131. https://doi.org/10.3390/cryst7050131
APA StyleDwyer, L., Kulkarni, S., Ruelas, L., & Myerson, A. (2017). Two-Stage Crystallizer Design for High Loading of Poorly Water-Soluble Pharmaceuticals in Porous Silica Matrices. Crystals, 7(5), 131. https://doi.org/10.3390/cryst7050131