1. Introduction
The computational modeling of materials has been a successful and rapid tool to address the unclear points of physical interest. In addition, predicting good elastic and thermodynamic properties of materials is a necessary demand for present-day solid state science and industry. In particular, these properties at high pressure and temperature are important for the development of modern technologies [
1,
2,
3,
4,
5].
As emphasized in the authors’ recent work, wide-band-gap II–VI semiconductors such as ZnX (X: S, O, Se, Te) and CdX (X: S, Se, Te) are remarkable materials for the design of high-performance opto-electronic devices including light-emitting diodes and laser diodes in the blue or ultraviolet region [
6].
According to its crystallographic structure, ZnS compounds can crystallize in either zinc blende (ZB) crystal structure with space group F-43m or wurtzite (WZ) crystal structure with the P6
3mc space group at ambient conditions [
7,
8,
9,
10].
To date, there have been a number of studies performed especially for the ground state (T = 0 K and P = 0 GPa) structural, elastic, thermodynamic, and optical properties of ZB and WZ crystal structures of ZnS compounds—not only with experiments, but also with theoretical works because of their technological importance. In 2004, Wright and Gale [
11] reported new interatomic potentials to model the structures and stabilities of the ZB and WZ ZnS phases in which their theoretical results are within experiments. Later, in 2008, Bilge et al. [
8], performed ab initio calculations based on projector augmented wave pseudo potential (PAW). They employed generalized gradient approximation (GGA) of density functional theory (DFT) to investigate the ground state mechanical and elastic properties of ZB, rock salt (NaCl), and WZ phases of ZnS. They concluded that the mechanical properties of ZnS under high pressure are quite different from those at ambient conditions. At the same time, Rong et al. [
10] reported the pressure dependence of the elastic properties of ZB and WZ crystals of ZnS by the GGA within the plane-wave pseudopotential (PWP) of DFT and found reasonable results which are consistent with former experimental and theoretical data. Further, in 2009, Cheng et al. [
12] conducted an experimental and theoretical study on first and second-order Raman scattering of ZB and WZ ZnS and found satisfactory experimental and theoretical results. Afterwards, in 2012, Grünwald et al. [
9] established transferable pair potentials for ZnS crystals to accurately describe the ground state properties of ZB and WZ phases of ZnS compounds. Recently, Yu et al. [
13] performed DFT calculations by using both the local density approximation (LDA) and GGA for the exchange-correlation potential and calculated the phonon dispersion curves and the phonon density of states of WZ ZnS in which the calculated values display good agreement with earlier experimental and theoretical data. In another recent study, Ferahtia et al. [
14] published the first-principles plane-wave-based pseudopotential method calculations of the structural, elastic, and piezoelectric parameters of WZ ZnS and concluded a reasonable degree of agreement between their results and data available in the literature.
The above recent and increasing theoretical efforts on ZnS motivated us to perform this work by addressing the lesser-known high-pressure elastic, mechanical, and phonon properties of WZ ZnS with a different method. In contrast to the above applied methods and interatomic potentials of literature, this is the first report regarding the application of a mixed-type potential to determine the mentioned high-pressure quantities of WZ ZnS with geometry optimization calculations. During calculations, we concentrated on the pressure behavior of five independent elastic constants, bulk, shear, and Young moduli, elastic wave velocities, mechanical stability conditions, and phonon properties of WZ ZnS under pressures between 0 GPa and 12 GPa at T = 0 K.
The following part of the article provides details of our theoretical calculations with the employed interatomic potential in
Section 2. In
Section 3, we compare our results with the available experimental results and other theoretical data. Finally,
Section 4 presents the main findings of this work in the conclusions.
2. Details of Theoretical Calculations
The most significant feature of materials modeling is the choice of the proper interatomic potentials that sufficiently and accurately describe the physical properties of the concerned problem. Simple empirical potentials are such modelling tools for materials, since they can yield reasonable results. These potentials can also provide good explanations of the defect energies, surface energies, elastic properties, and mechanical aspects of oxides [
15], fluorides [
16] and many other materials [
17,
18]. Most of these potentials include Coulomb interactions, short-range pair interactions, and ionic polarization treated by the shell model of Dick and Overhauser [
19]. The sum of the Coulomb terms, short-range interactions, and ionic polarization expresses the total energy for these potentials. If we presume that the electron cloud of an ion is simulated by a massless shell charge
qs and the nucleus by a core of charge
qc, then the total charge becomes
q =
qs +
qc. In the shell model interatomic potential approach, a harmonic force with spring constant
couples the core and the shell of the ion. So, for modelling the short-range pair interactions acting between the shells, we can use a typical Buckingham potential, as presented in Equation (1):
The first part of Equation (1) indicates the Born–Mayer term, whereas the second part represents the Van der Waals energies. Further, in our work, we applied the original form of a mixed-type interatomic potential of Hamad et al. [
20] for short-range interactions, which incorporates the Buckingham and Lennard–Jones 9–6 potentials form as in Equation (2):
We also considered the semi-covalent nature of ZnS by using a three-body potential for S-Zn-S angel as in its original form [
20] and expressed by Equation (3):
In Equation (3),
and
show the equilibrium constant angle between S-Zn-S and fitting constant of Hamad et al. [
20], respectively. Lastly, sulphur anion polarizibility treated by the shell model of Reference [
19] can be written as in Equation (4):
where
describes the core–shell separation, and
indicates the spring constant. Although extra details of presently employed potential and its parameterization procedure can be found in Reference [
20],
Table 1 lists the original potential parameters of Reference [
20] used in our calculations.
All theoretical calculations in this work were carried out with the General Utility Lattice Program (GULP) 4.2 molecular dynamics code [
21,
22]. This versatile code allows the concerned structures to be optimized at constant pressure (all internal and cell variables are included) or at constant volume (unit cell remains frozen). To avoid the constraints, constant pressure optimization was applied to the geometry of WZ ZnS cell with the Newton–Raphson method based on the Hessian matrix calculated from the second derivatives. The cell geometry of WZ ZnS was assigned as
, u = 0.375, and
and
with space group
. During the present geometry optimization calculations, the Hessian matrix was recursively updated with the BFGS [
23,
24,
25,
26] algorithm. After setting the necessities for the geometry optimization of WZ ZnS, we devised multiple runs at zero Kelvin temperature and checked the pressure ranges between 0 GPa and 12 GPa in steps of 3 GPa. Further, phonon and associated properties of WZ ZnS were also addressed after constant pressure geometry optimization calculations as a function of pressure within the quasiharmonic approximation under zero Kelvin temperature, as implemented in GULP code. It is possible to capture the phonon density of states (PDOS) and dispersions for a material after specifying a shrinking factor with GULP phonon computations. Further, phonons are described by calculating their values at points in reciprocal space within the first Brillouin zone of the given crystal. To achieve the Brillouin zone integration and obtain the PDOS, we have used a standard and reliable scheme developed by Monkhorts and Pack [
27] with 8 × 8 × 8
k-point mesh.
3. Results and Discussion
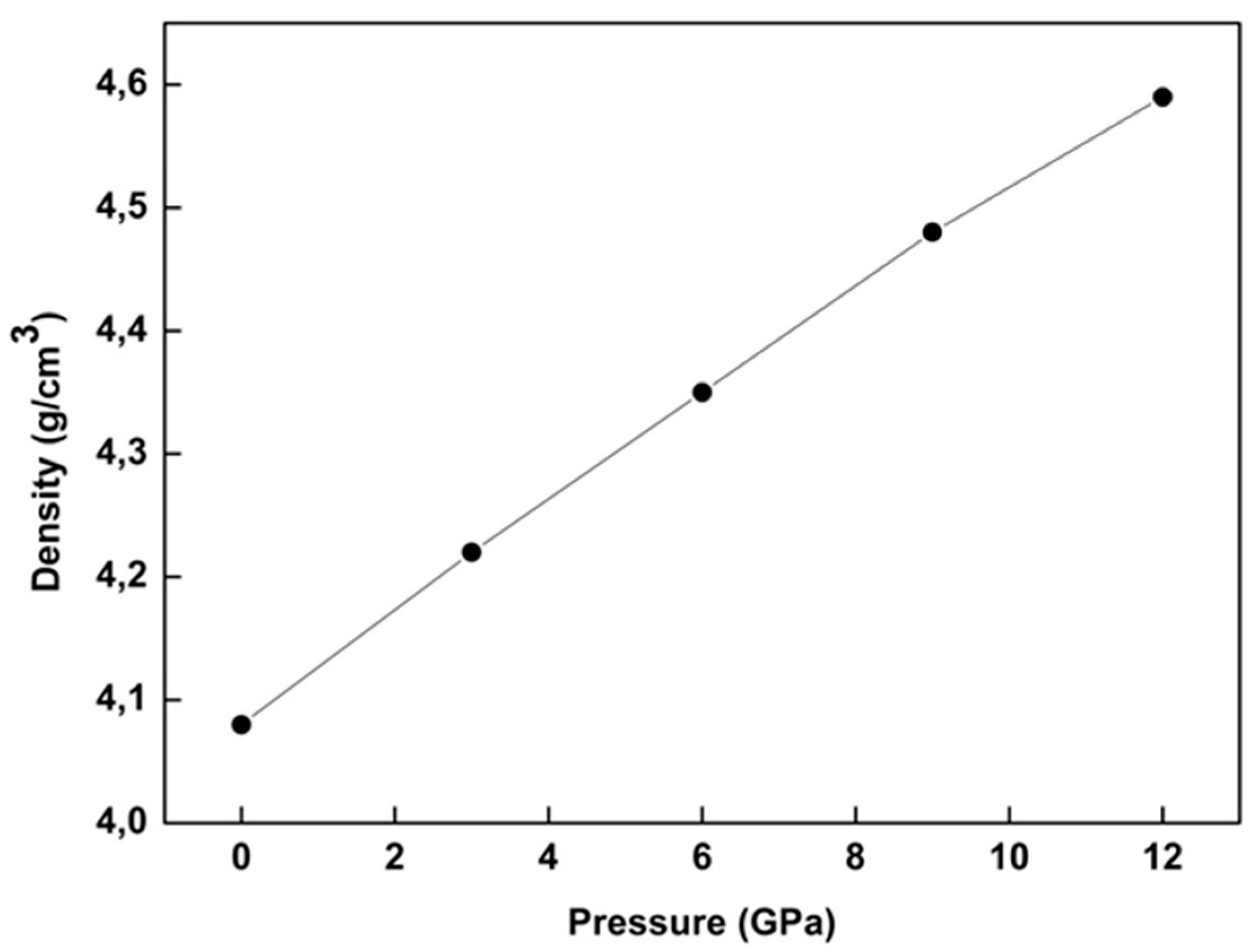
Figure 1 indicates the density behavior of WZ ZnS under pressure. As is well known, the density of many materials exhibits clear increments under pressure [
6,
15,
17,
18,
28]. This is also the case for WZ ZnS under pressure, as seen in
Figure 1. The minimum and maximum values of the density of WZ ZnS was found to be 4.08 g/cm
3 and 4.59 g/cm
3 for 0 GPa and 12 GPa, respectively, at zero temperature.
The elastic constants of materials not only provide precise and essential information about the materials, but also explain many mechanical and physical properties. Once the elastic constants are determined, one may get a deeper insight into the stability of the concerned material [
6,
15,
17,
18,
28]. These constants can be also helpful to predict the properties of materials (i.e., interatomic bonding, equation of state, and phonon spectra). They also link to several thermodynamic parameters, such as the specific heat, thermal expansion, Debye temperature, Grüniesen parameter, etc. However, in general, elastic constants derived from the total energy calculations correspond to single-crystal elastic properties. So, the Voigt–Reuss–Hill approximation is a confident scheme for polycrystalline materials [
6,
15,
17,
18,
28]. To obtain the accurate values of elastic constants and other analyzed parameters of WZ ZnS, the Voigt–Reuss–Hill values were taken into account for this research. For wurtzite crystal structures, five independent elastic constants exist as:
[
28].
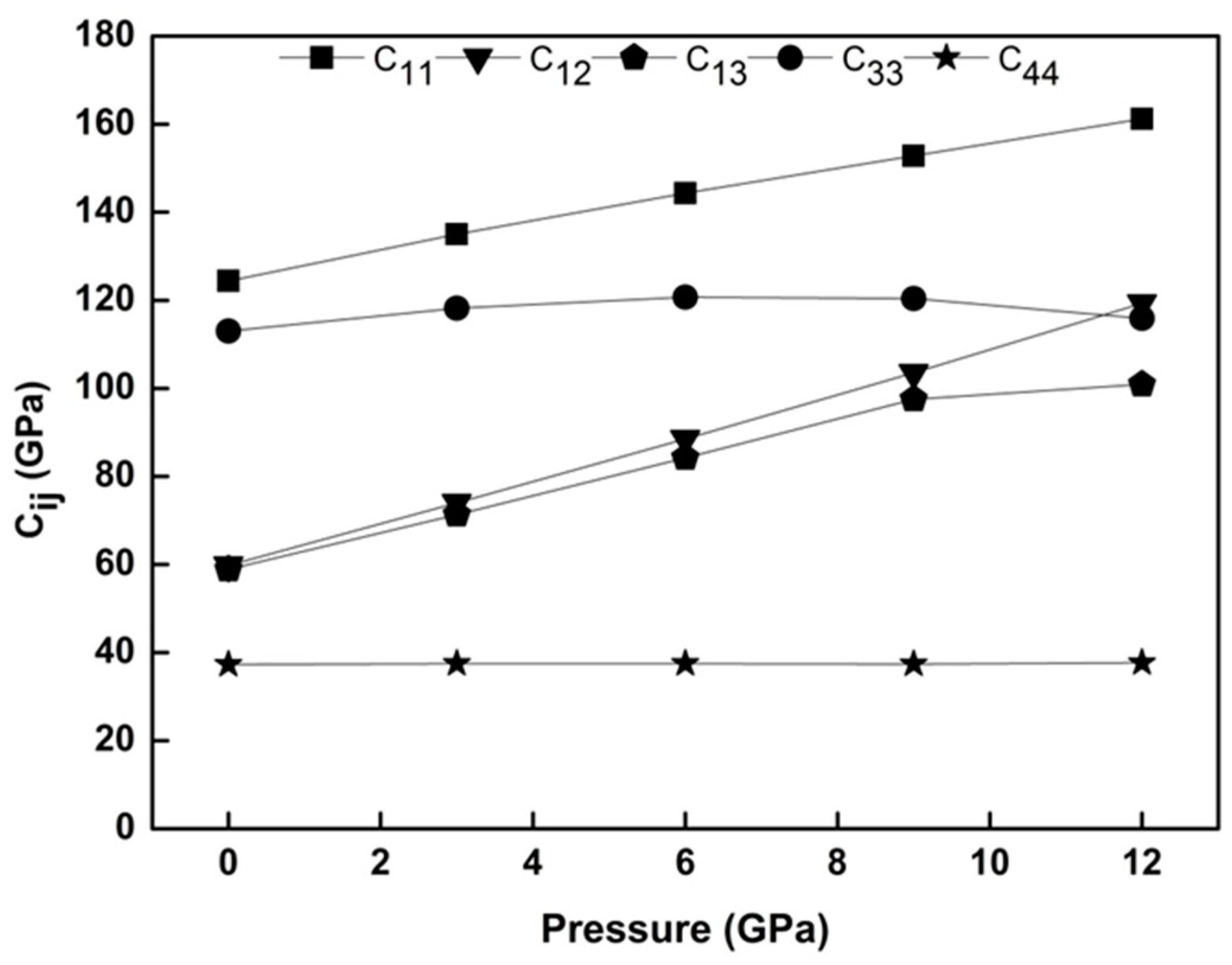
The plot in
Figure 2 shows our results for
elastic constants of WZ ZnS under pressures between 0 GPa and 12 GPa. All obtained elastic constants of WZ ZnS increase with the applied pressure except
. Beyond these increments, a closer examination of
Figure 2 reveals that the magnitudes of the elastic constants are in the range of:
>
>
>
>
. Both the range of elastic constants and slight decrement of
constant under pressure mimic the DFT findings of Tan et al. [
7]. However, our results for the ground state parameters of WZ ZnS are obviously better than those of Tan et al. [
7] as well as Wright and Gale [
11], and are much closer to the experimental results of Neumann et al. [
29].
Table 2 gives a numerical comparison of present results for the elastic constants and other calculated quantities of WZ ZnS with some previously published experimental and theoretical data.
From a stability outlook, the proverbial Born mechanical stability condition for a hexagonal structure also holds for wurtzite crystal, and must satisfy [
28]:
The present results for the obtained elastic constants of WZ ZnS satisfy the mechanical stability condition (
Figure 2 and
Table 2), and this result points out that WZ ZnS is mechanically stable at 0 K temperature and 0 GPa pressure.
The bulk modulus (
B) is the only elastic constant of a material that conveys much information about the bonding strength. Moreover, it is a measure of the material’s resistance to external deformation, and occurs in many formulae describing diverse mechanical–physical characteristics. The shear modulus (
G), however, portrays the resistance to shape change caused by a shearing force. In addition to
B and
G, Young’s modulus (
E) is the resistance to uniaxial tensions. These three distinct moduli (
B,
G, and
E) are other valuable parameters for identifying the mechanical properties of materials [
6,
15,
17,
18,
28].
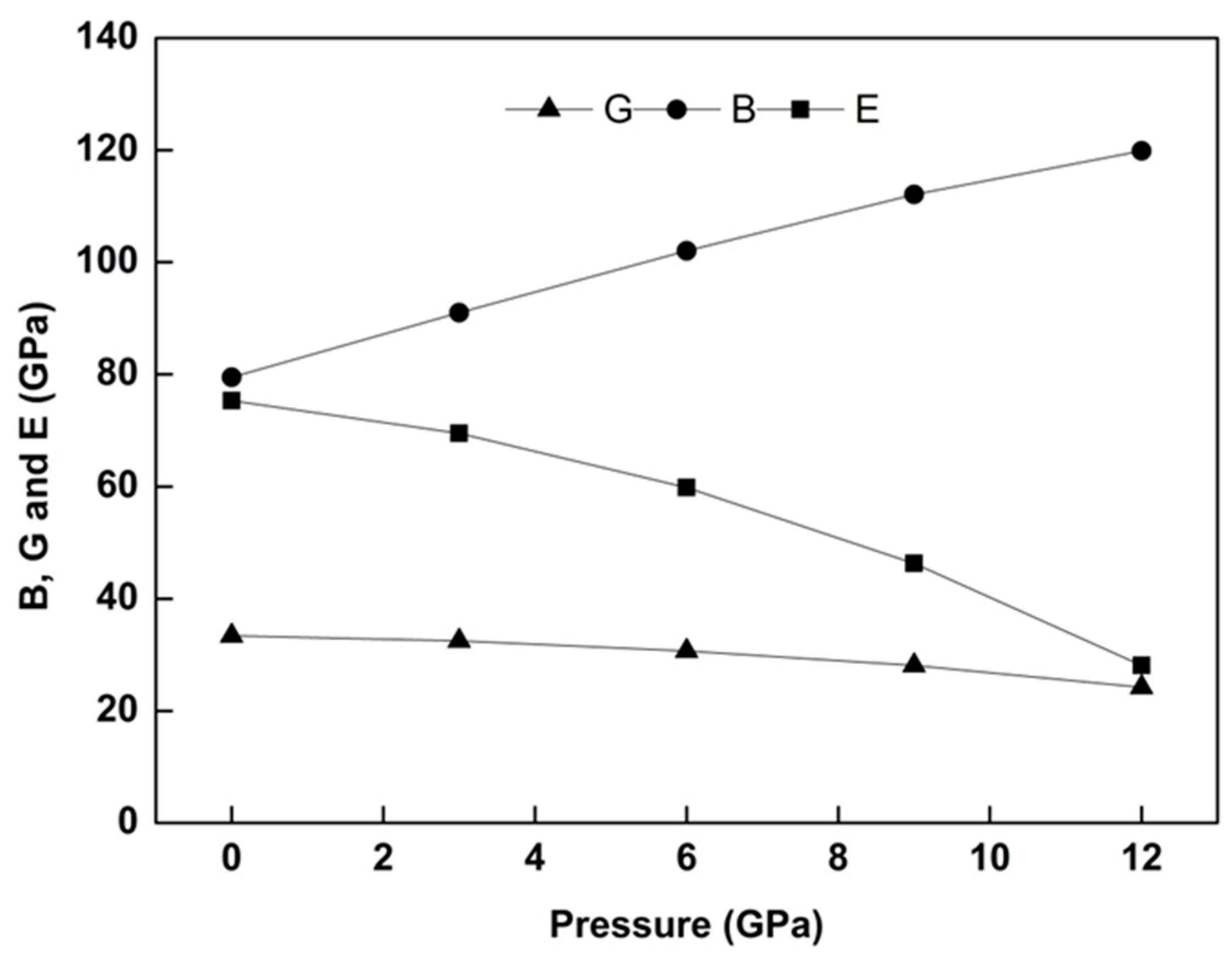
Figure 3 represents the pressure behavior of
of WZ ZnS for the entire pressure range. From the prevalent physical definition of bulk modulus
is expected an increment for
because of its direct proportion to applied pressure. Hence, bulk modulus of WZ ZnS exemplifies a straight increment in
Figure 3. Contrary to sharp increment in B, E and G moduli have slight decrements under pressure again similar to the DFT findings of Tan et al. [
7]. Apart from the pressure behavior of these three moduli,
Table 2 also summarizes another numerical comparison for
with former experimental and theoretical data. The present result for the bulk modulus of WZ ZnS with 79.5 GPa approximately agrees with experimental values and is better than other DFT results, as seen in
Table 2.
The adjectives “brittle” and “ductile” signify the two distinct mechanical characters of solids when they are exposed to stress. Ductile and brittle features of materials play a key role during the manufacturing of materials [
6,
15,
17,
18,
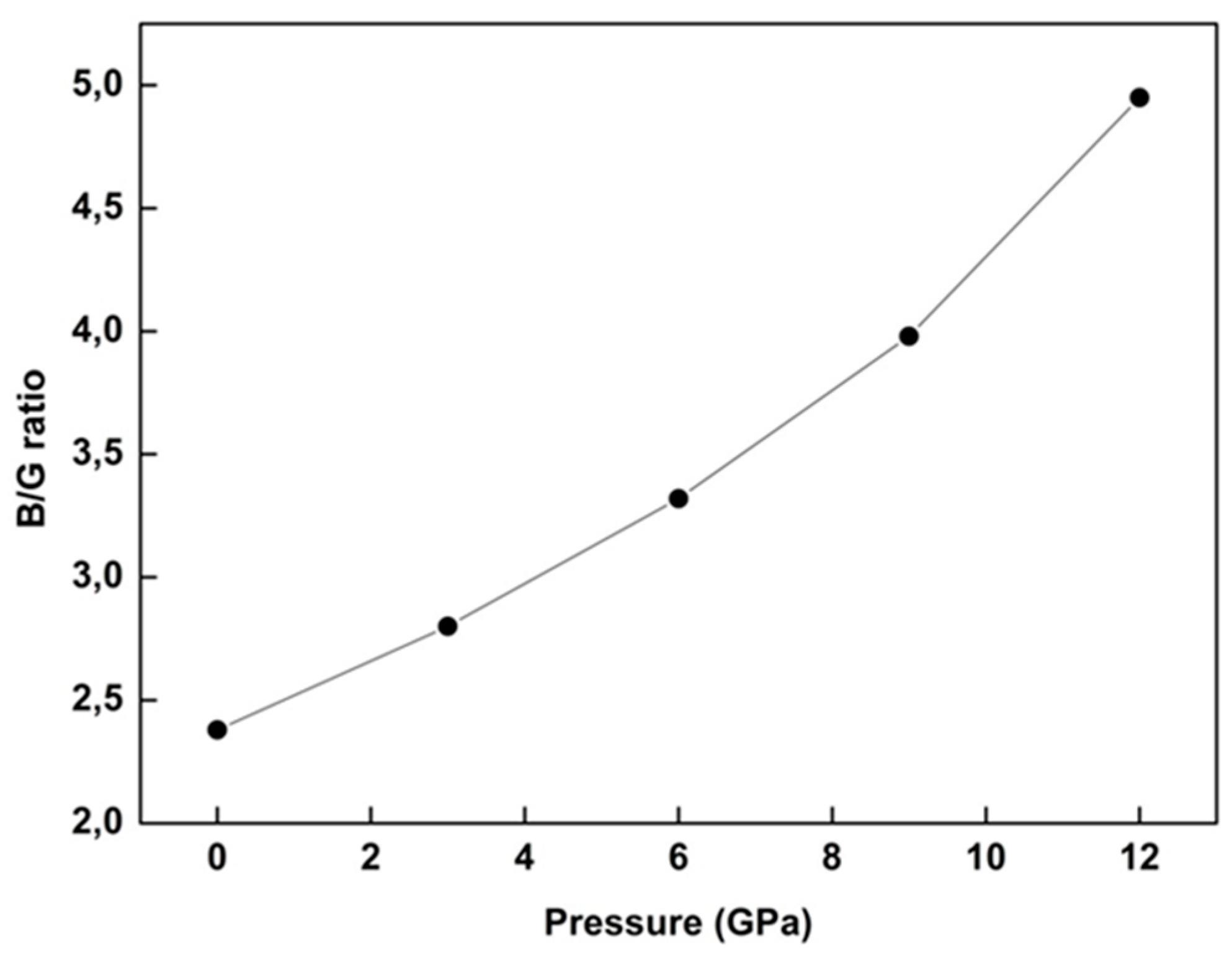
28]. So, we also evaluated the ductile (brittle) nature of WZ ZnS under pressure. In general, brittle materials are not deformable or are less deformable before fracture, whereas ductile materials are very deformable before fracture. At this point, the Pugh ratio is a determinative limit for the ductile (brittle) behavior of materials, and has popular use in literature. If the B/G ratio is about 1.75 and higher, the material is accepted to be ductile; otherwise, the material becomes brittle [
6,
15,
17,
18,
28]. After a careful evaluation, we determined B/G values changing from 2.38 to 4.95, with a monotonous increment between P = 0 GPa and P = 12 GPa at zero temperature, respectively, for WZ ZnS (as seen in
Figure 4). So, this result manifests the ductile character of WZ ZnS in both ground state and under pressure. As another result, all values of the B/G are higher than 1.75 and increase with pressure, which suggests that pressure can improve ductility (
Figure 4).
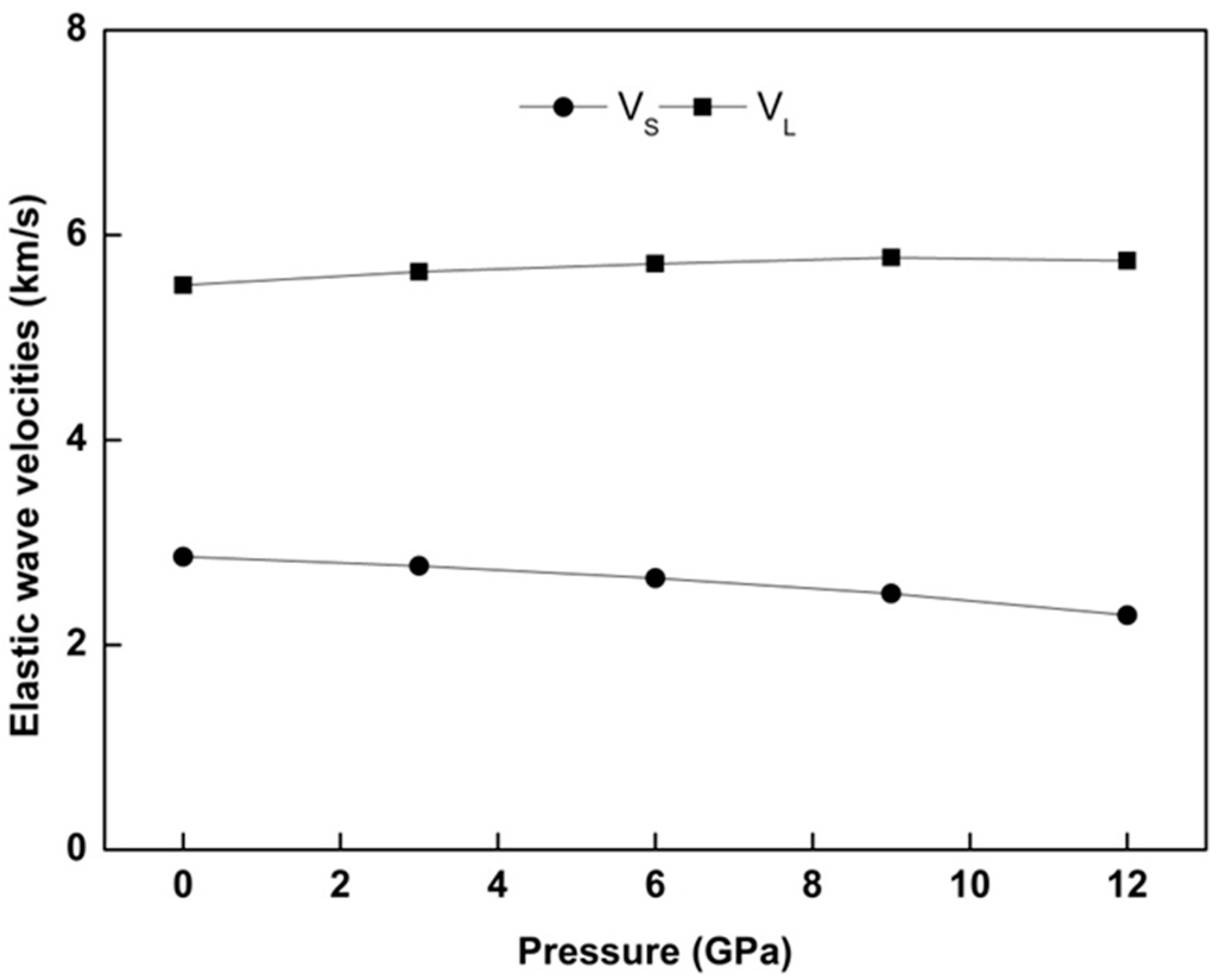
In solids, low temperature (T = 0 K in our case) acoustic modes can trigger the vibrational excitations. Depending on this fact, two typical (longitudinal and shear) elastic waves exist. The velocity
represents the longitudinal elastic wave velocity and
denotes shear wave velocity [
6,
15,
17,
18,
28].
Figure 5 shows the behavior of
and
elastic wave velocities for WZ ZnS as a function of pressure.
has a substantial increment compared to
, which is the most observed circumstance for many materials.
Figure 6 shows the phonon dispersion of WZ ZnS along the chosen Γ–A path. In addition,
Table 3 compares the numerical values of the zone–center (Γ points) phonon frequencies of this work with earlier experimental and theoretical data of WZ ZnS. As seen in
Table 3, the agreement with experiment is quite good, especially for A1 (TO), E1 (TO) and A2 (TO), E2 (TO) phonon modes and competing with recent GGA and LDA DFT data of Reference [
14].
Further, we have focused on the ground state phonon density of states (PDOS) of WZ ZnS to clarify the contribution of the both elements (Zn and S) to the phonon properties of the compound.
Figure 7 demonstrates the partial PDOS of WZ ZnS under zero pressure and temperature. As is apparent in
Figure 7, the contribution of the Zn element to acoustic phonon modes is higher than S, whereas the opposite case is valid for the S element due to its dominant contribution to the optical modes of the compound.
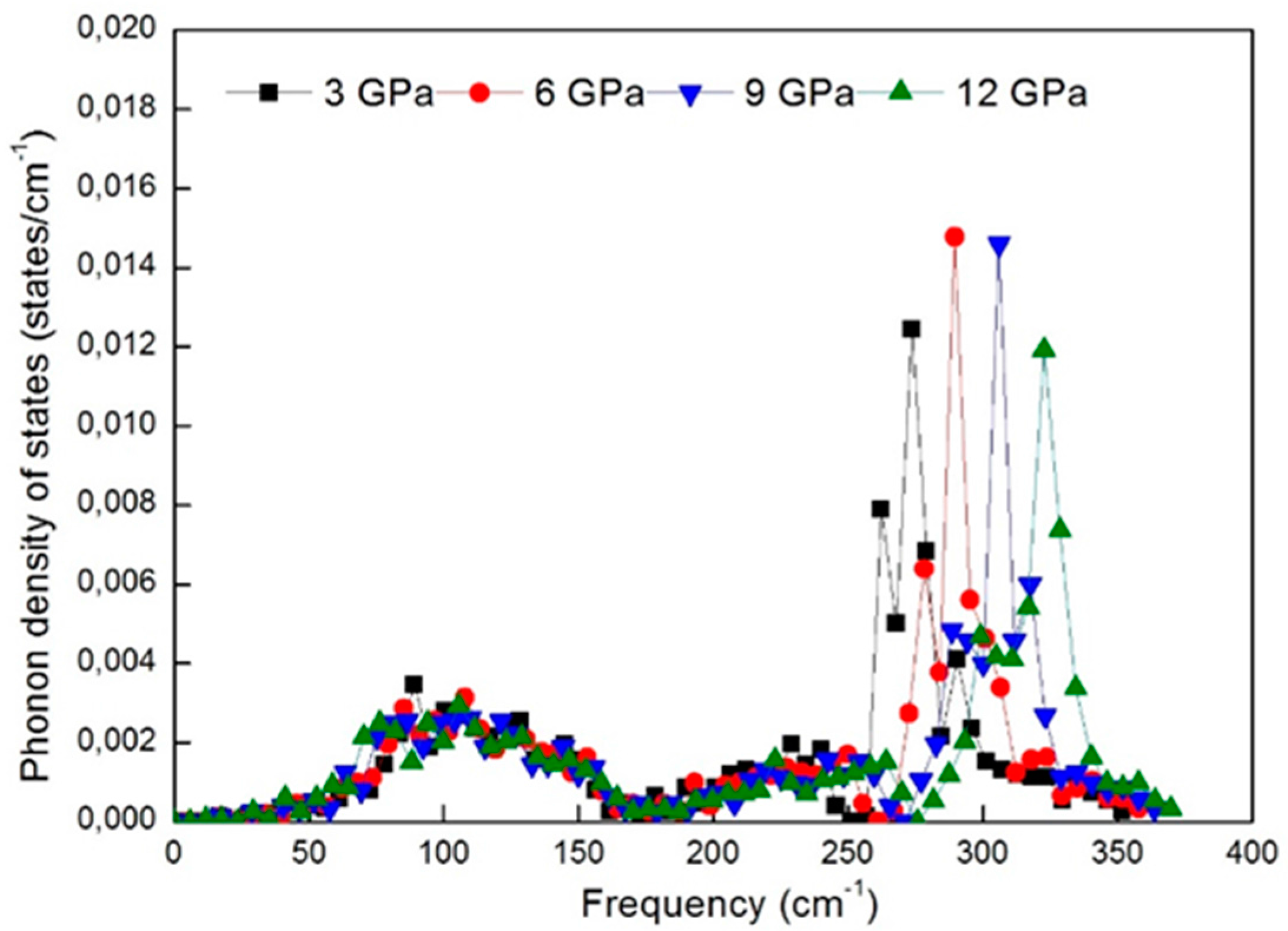
Figure 8 shows the phonon dispersion curves of WZ ZnS under different pressures. Each pressure value above 0 GPa shifts the dispersion curves of WZ ZnS up slightly to the higher frequency values, as is clear in
Figure 8. This behavior strictly originates from the atoms of WZ ZnS which are getting closer to each other under pressure, since they sit in steeper potential wells. The effect of pressure also causes the same behavior in the total density of states curves of WZ ZnS as shown in
Figure 9.
Overall, the obtained results display a fair agreement with the experiments—in particular in elastic constants, bulk modulus, and phonon properties of WZ ZnS. Finally, the presented results for all considered quantities of WZ ZnS through this research are not only consistent with experiments, but also better than those of some published theoretical data.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}