A Novel Approach to Data Collection for Difficult Structures: Data Management for Large Numbers of Crystals with the BLEND Software

 and
and
Abstract
:1. Introduction
- Increased likelihood of solving a structure even before having obtained well-diffracting crystals through the use of optimised crystallization conditions. This amounts to a significant saving in time, as many attempts are very often needed to find the right conditions that yield large crystals.
- Increased data multiplicity, with the twofold consequence of obtaining better data scaling and stronger anomalous signal, if strong anomalous scatterers are present in the structure. A consequence of this so-called data redundancy is the recent finding that native proteins can be solved by exploiting the generally faint anomalous signal due to sulphur atoms, because such a signal is highly enhanced by the high data multiplicity [7,8,10,14,16,17].
- More accurate structure factors. As scaled data are obtained merging individual observations from different, independent crystals, the derived structure factors might present larger errors, but better accuracy. Phasing and the resulting electron density maps, accordingly, have improved overall quality [3]. This qualitative observation holds if the different crystals have a reasonable level of isomorphism.
- Physical limitation of the deteriorating effects due to radiation damage. Only the first portion of every dataset can be retained when merging data together, because later sweeps generally include reflections biased by the changing lattice, progressively altered by X-ray radiation.
- New scenarios opened by the management of multiple datasets in relation to crystals isomorphism and structure dynamics. One such scenario is the use of multiple crystals for structure-guided drug design, whereby many crystals are soaked in a cocktail of chemical fragments that act as precursors for more complex drug molecules. Data is then collected from multiple crystals and merged to produce electron density maps that allow the identification of bound inhibitors [18,19].
Datasets from Single and Multiple Crystals
2. Collecting Data from More Than One Crystal: A Short Review
3. The BLEND Program
The Absolute Linear Cell Variation (aLCV)
4. Materials and Methods
4.1. The Target Structure
4.2. Data Collection and Plans to Solve the Structure
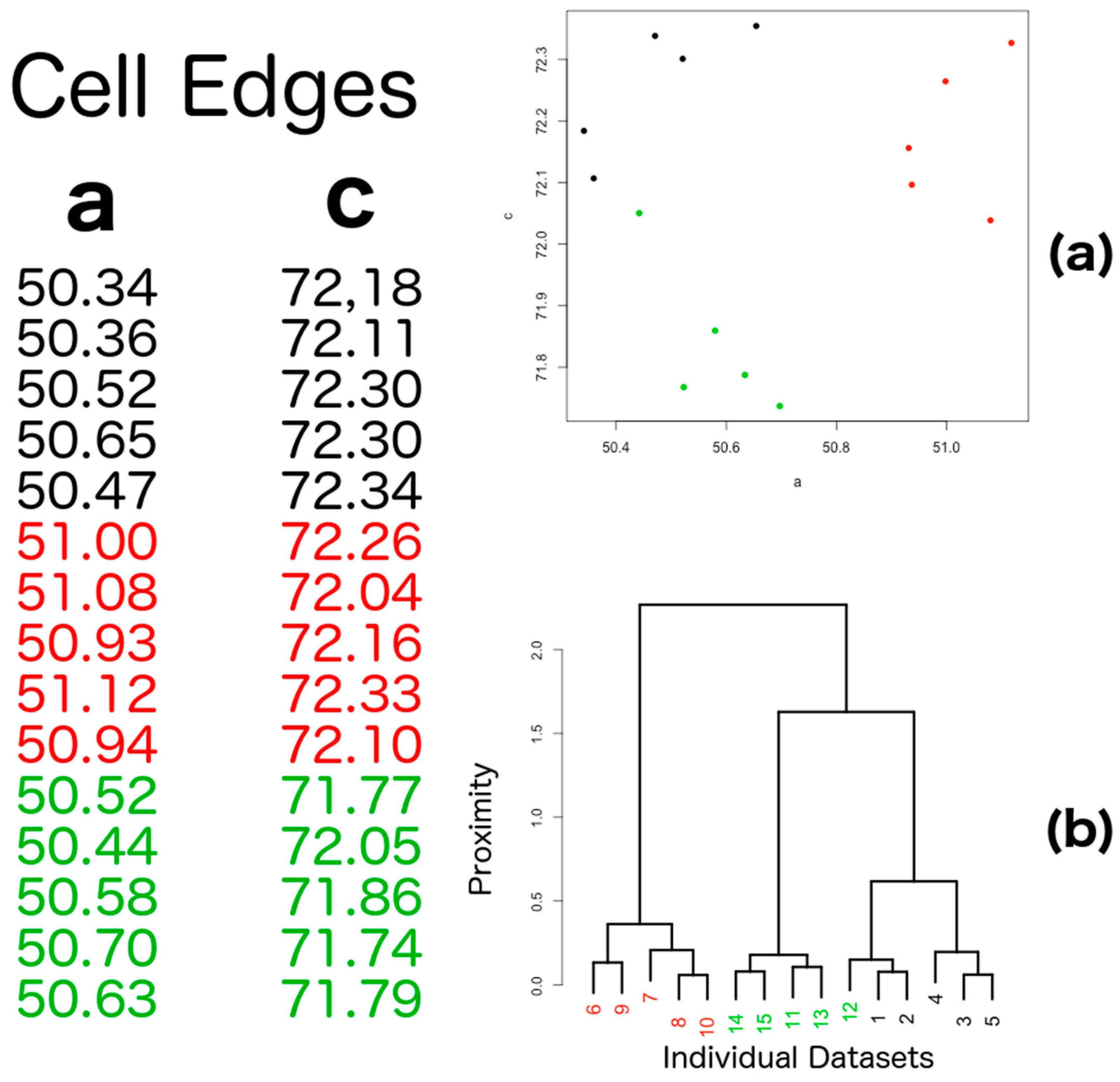
4.3. Pre-Clustering
4.4. Strategy for Data Combination
4.5. Detailed Description
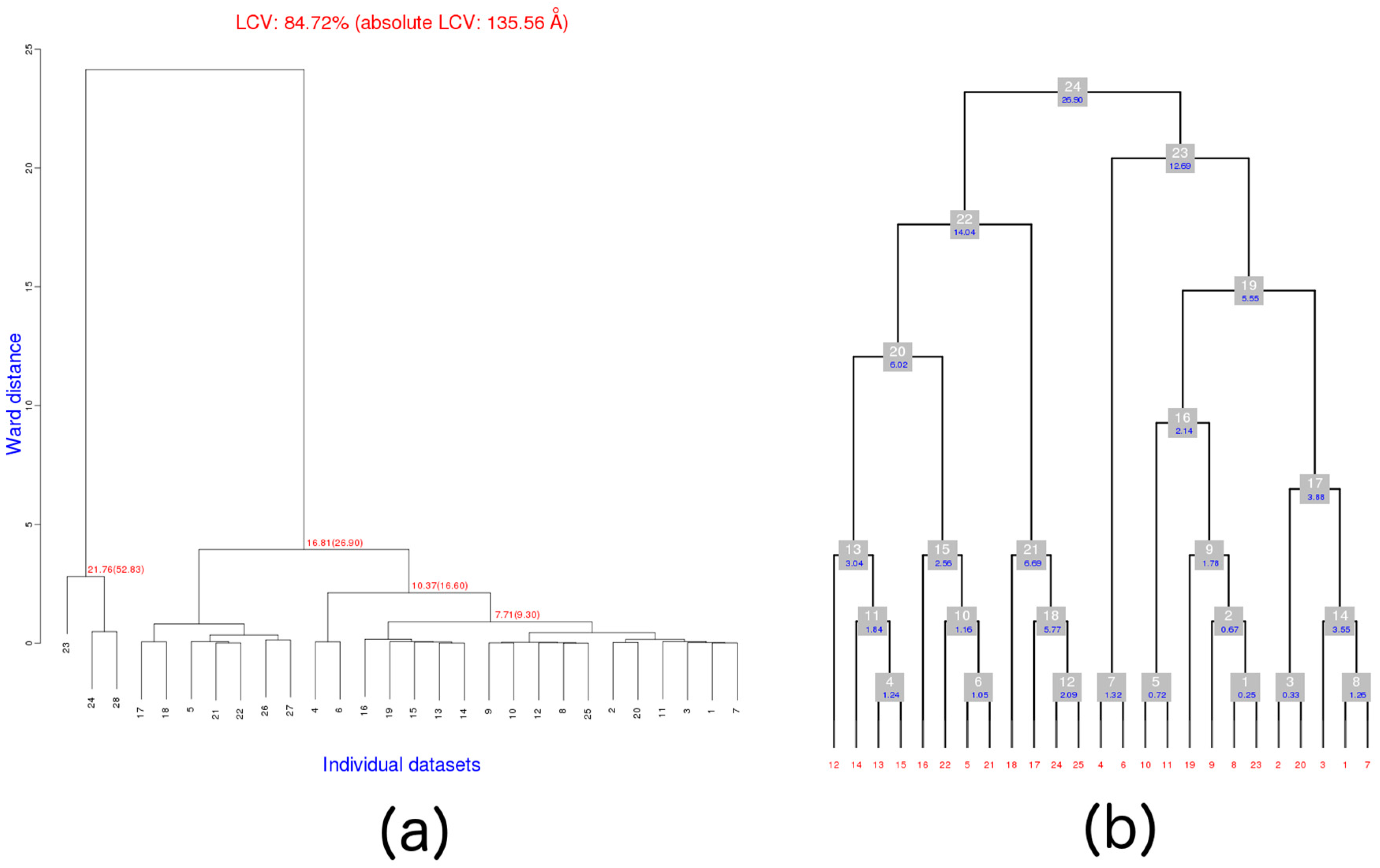
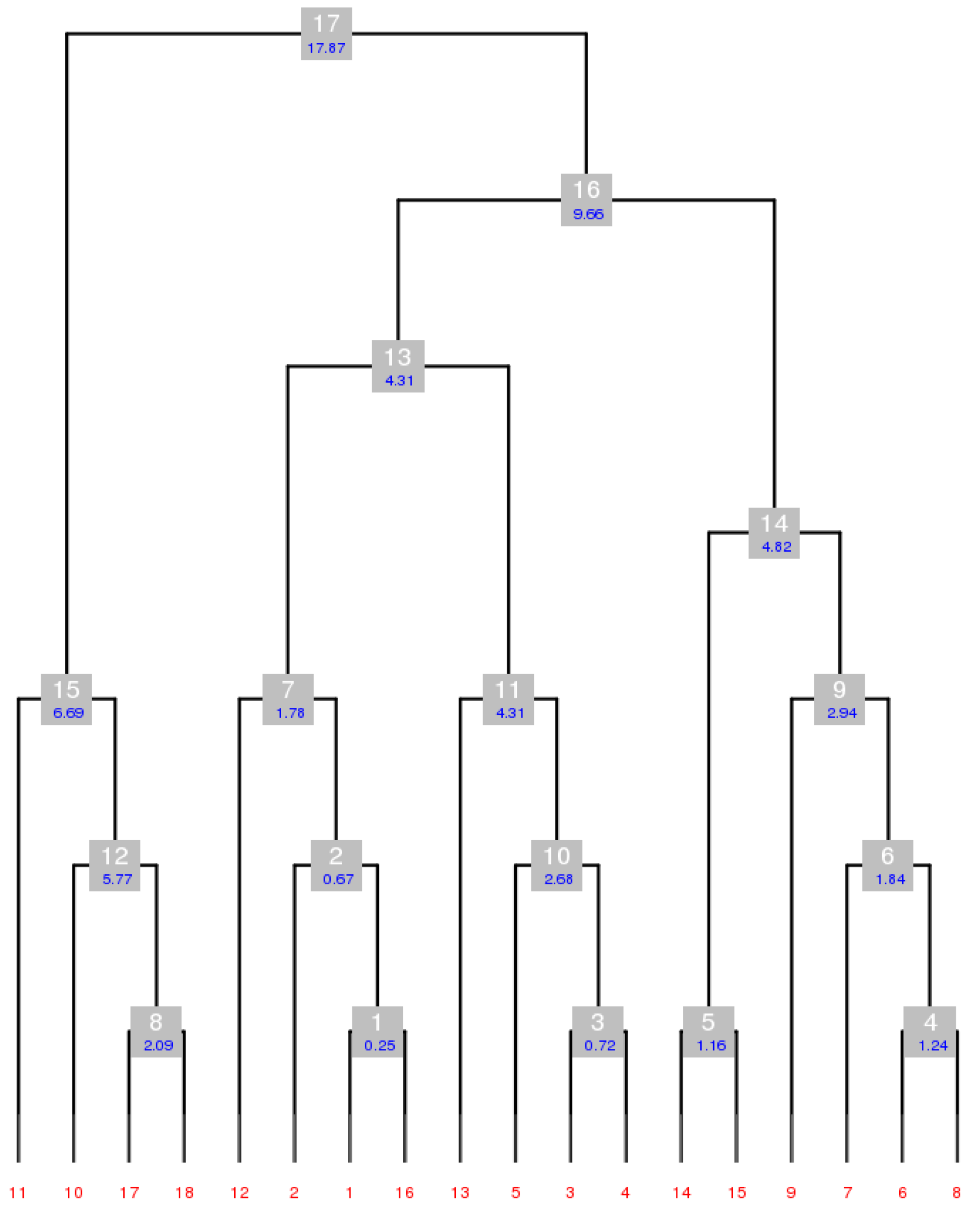
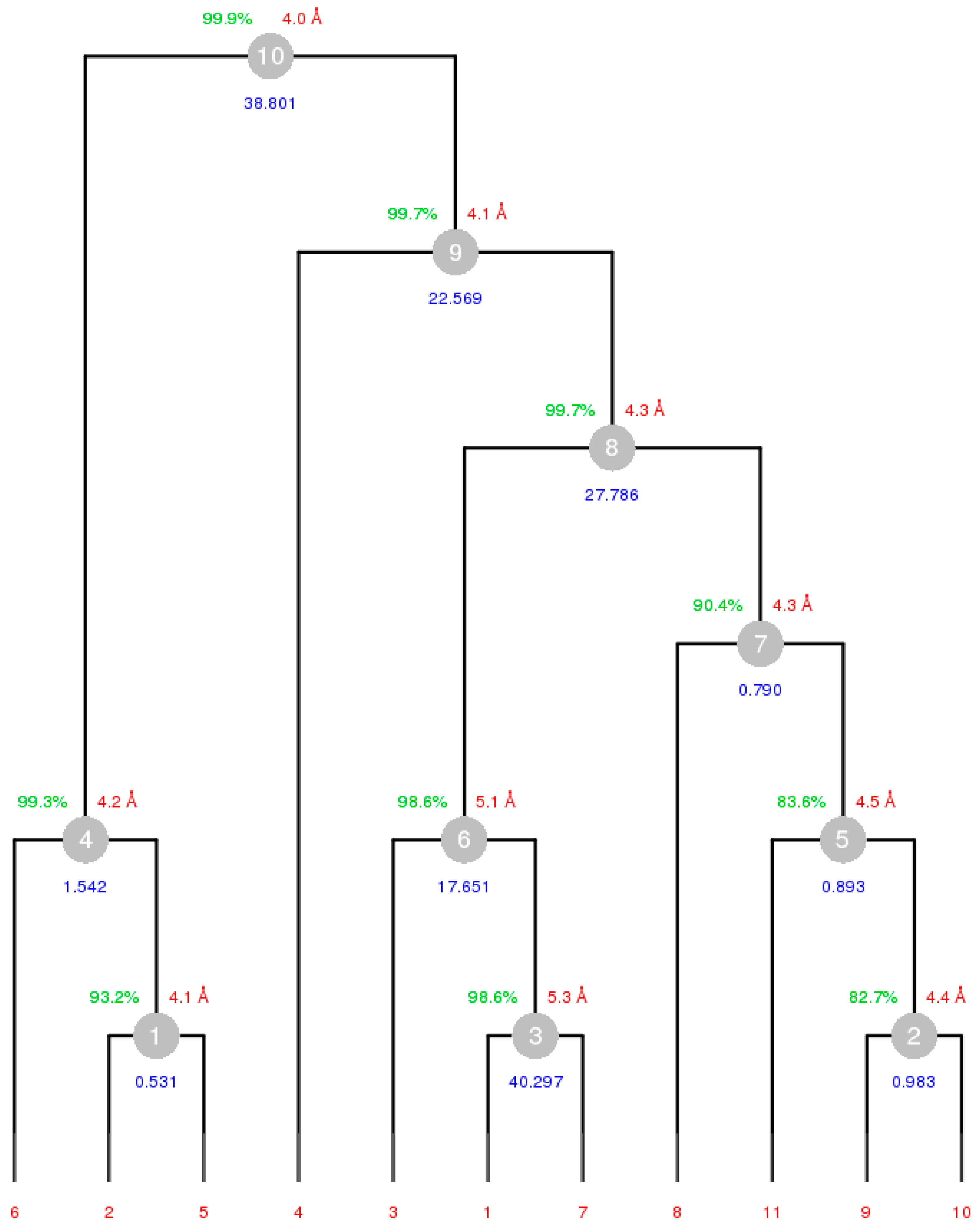
4.5.1. Working out DMCs with Serial Group 25
4.5.2. Obtaining DMCs from Serial Group 2
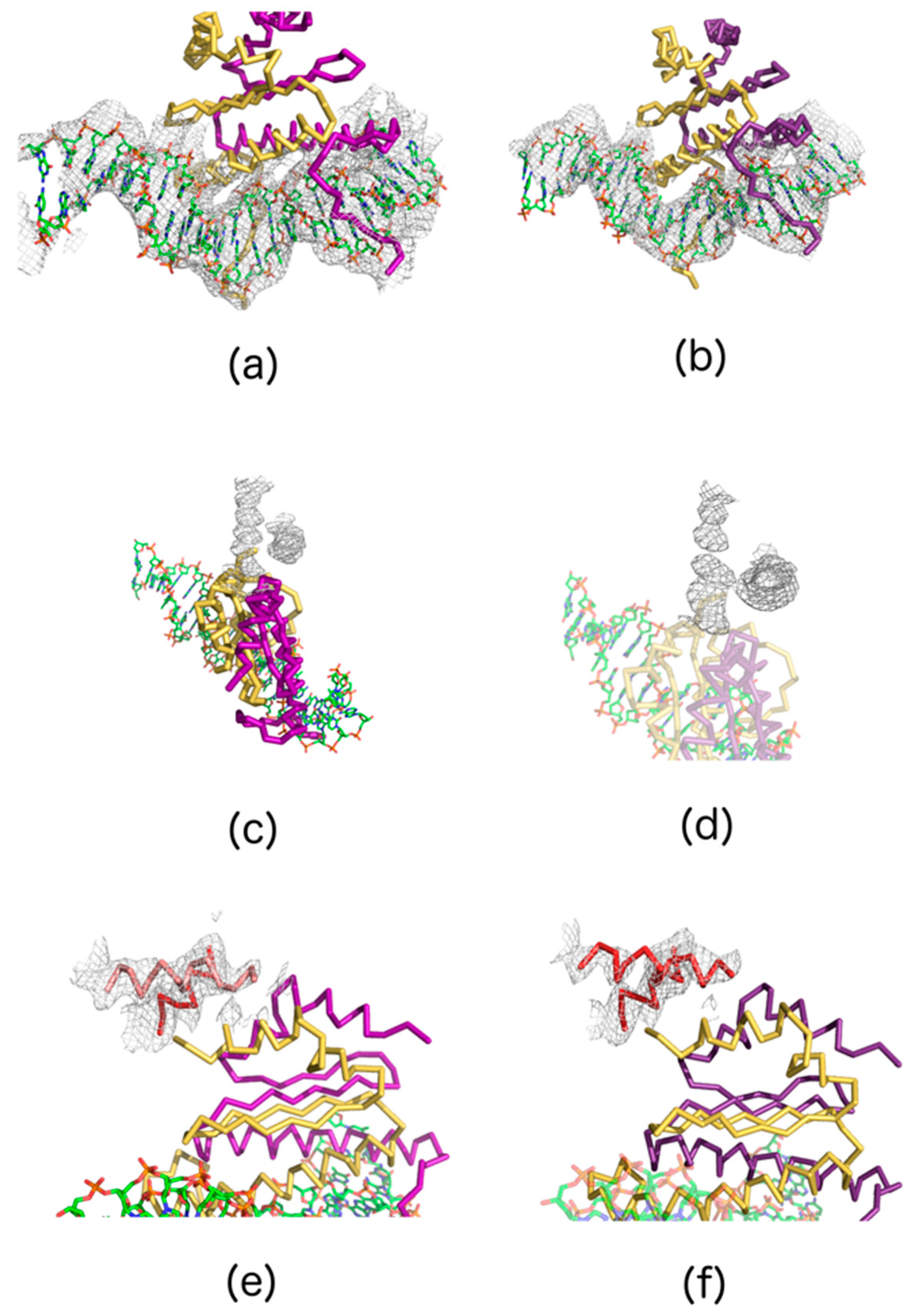
4.6. Structure Solution
4.6.1. Data Used
4.6.2. Molecular Replacement
4.6.3. Structure Refinement and Electron Density
5. Discussion and Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
- (1)
- bc1 = A + B + C1
- (2)
- bc2 = A + B + C2
- (3)
- bc3 = A + B + C1 + Gd
- A = commercial screen
- B = commercial screen for optimization
- C1 = additive screen
- C2 = additive screen
- Gd = gadolinium
- (1)
- cry1 = 30% glycerol + 5 mM magnesium chloride
- (2)
- cry2 = 30% glycerol + OH
- (3)
- cry3 = 30% glycerol + 5 mM magnesium chloride + 1 M sodium bromide

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
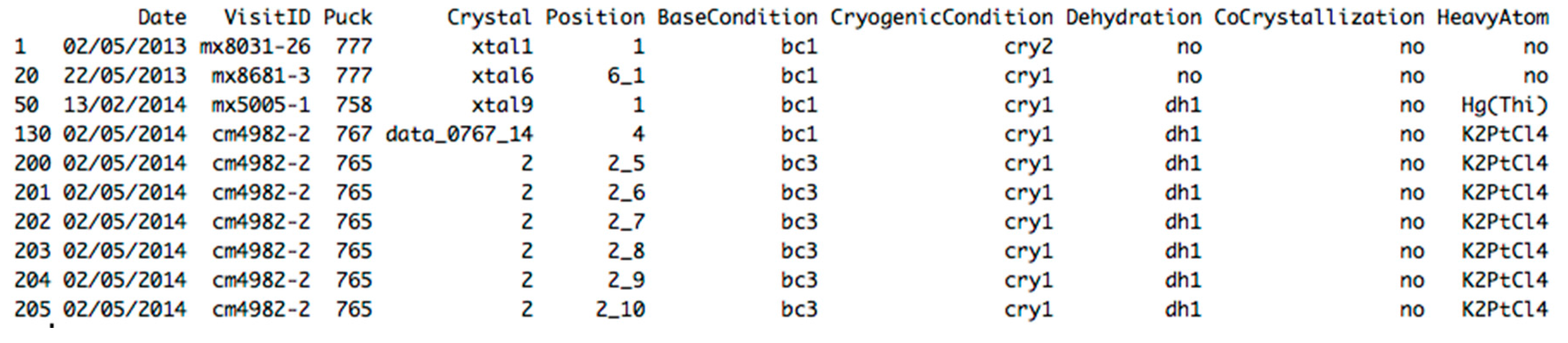
| Date | Visit ID | Puck | Crystal | Position | Serial N. | Base Condition | Cryogenic Condition | Dehydration | Co-Crystallized | Heavy Atom |
|---|---|---|---|---|---|---|---|---|---|---|
| 02/05/2013 | mx8031-26 | 777 | xtal1 | 1 | 1 | bc1 | cry2 | no | no | no |
| 2 | 2 | |||||||||
| 3 | 3 | |||||||||
| 4 | 4 | |||||||||
| xtal3 | 1 | 5 | bc1 | cry2 | no | no | no | |||
| 2 | 6 | |||||||||
| 3 | 7 | |||||||||
| 4 | 8 | |||||||||
| 5 | 9 | |||||||||
| xtal6 | 1 | 10 | bc1 | cry2 | no | no | no | |||
| 2 | 11 | |||||||||
| 3 | 12 | |||||||||
| xtal8 | 1 | 13 | bc1 | cry2 | no | no | no | |||
| b_1 | 14 | |||||||||
| 22/05/2013 | mx8681-3 | 777 | xtal3 | real_3_2 | 15 | bc1 | cry1 | no | no | no |
| real_3c_2 | 16 | |||||||||
| real_3d_3 | 17 | |||||||||
| real_3e_2 | 18 | |||||||||
| xtal4 | 1 | 19 | bc1 | cry1 | no | no | no | |||
| xtal6 | 6_1 | 20 | bc1 | cry1 | no | no | no | |||
| 6a_2 | 21 | |||||||||
| 6b_1 | 22 | |||||||||
| 6b_2 | 23 | |||||||||
| 6c_1 | 24 | |||||||||
| xtal9 | 9_1 | 25 | bc2 | cry1 | no | no | no | |||
| 9b_1 | 26 | |||||||||
| 9b_2 | 27 | |||||||||
| 9b_3 | 28 | |||||||||
| xtal14 | b_1 | 29 | bc2 | cry1 | no | no | no | |||
| 778 | xtal3 | 3_2 | 30 | ? | ? | ? | ? | ? | ||
| 20/10/2013 | mx8681-13 | 542 | 1_13 | 3 | 31 | bc1 | cry1 | dh1 | no | no |
| 5 | 32 | |||||||||
| 6 | 33 | |||||||||
| 7 | 34 | |||||||||
| 8 | 35 | |||||||||
| 9 | 36 | |||||||||
| 10 | 37 | |||||||||
| 13/02/2014 | mx5005-1 | 136 | 7 | 7 | 38 | bc1 | cry1 | dh1 | no | KICl6 |
| 7b | 39 | |||||||||
| 8 | 8 | 40 | bc1 | cry1 | dh1 | no | KICl6 | |||
| 8b | 41 | |||||||||
| 10 | 10 | 42 | bc1 | cry1 | dh1 | no | KICl6 | |||
| 10b | 43 | |||||||||
| 138 | xtal15 | 15_1 | 44 | bc1 | cry1 | dh1 | no | Tantalum | ||
| 542 | xtal2 | 1 | 45 | bc1 | cry1 | dh1 | no | Hg (Thi) | ||
| xtal4 | 1 | 46 | bc1 | cry1 | dh1 | no | Hg (Thi) | |||
| 544 | xtal2 | _ | 47 | bc1 | cry1 | dh1 | no | Pt (PIP) | ||
| 546 | xtal4 | 4_1 | 48 | bc1 | cry1 | dh1 | yes | KAu(CN)2 | ||
| 758 | xtal1 | 1 | 49 | bc1 | cry1 | dh1 | no | Hg (Ace) | ||
| xtal9 | 1 | 50 | bc1 | cry1 | dh1 | no | Hg (Thi) | |||
| 762 | xtal1 | 2 | 51 | bc1 | cry1 | dh1 | no | K2PtCl4 | ||
| xtal2 | data | 52 | bc1 | cry1 | dh1 | no | K2PtCl4 | |||
| xtal4 | 1 | 53 | bc1 | cry1 | dh1 | no | K2PtCl4 | |||
| xtal13 | 1 | 54 | bc1 | cry1 | dh1 | no | KAu(CN)2 | |||
| xtal14 | 1 | 55 | bc1 | cry1 | dh1 | no | KAu(CN)2 | |||
| xtal15 | 1 | 56 | bc1 | cry1 | dh1 | no | KAu(CN)2 | |||
| 764 | xtal14 | 3 | 57 | bc1 | cry1 | no | no | no | ||
| 4 | 58 | |||||||||
| 5 | 59 | |||||||||
| 765 | xtal5 | 5_1 | 60 | bc1 | cry1 | dh1 | no | Hg (PMA) | ||
| 766 | xtal3 | 3_1 | 61 | bc1 | cry1 | dh1 | no | K2PtI6 | ||
| 17/02/2014 | cm4982-1 | CPS-0134 | 12 | 2 | 62 | bc1 | cry1 | dh1 | yes | OsCl3 |
| 13 | 2 | 63 | bc1 | cry1 | dh1 | yes | K2PtCl4 | |||
| 3 | 64 | |||||||||
| 4 | 65 | |||||||||
| CPS-0140 | 7 | 7_1 | 66 | bc1 | cry1 | dh1 | yes | K2PtCl4 | ||
| 11 | 11_4 | 67 | bc1 | cry1 | dh1 | yes | Pt (PIP) | |||
| 12 | 12_1 | 68 | bc1 | cry1 | dh1 | yes | AgN | |||
| 15 | 15_1 | 69 | bc1 | cry1 | dh1 | yes | I3C (m.triangle) | |||
| CPS-0761 | 1 | 2 | 70 | bc1 | cry1 | dh1 | yes | GdCl3 | ||
| 2 | 2 | 71 | bc1 | cry1 | dh1 | yes | GdCl3 | |||
| line | 72 | |||||||||
| 5 | 1 | 73 | bc1 | cry1 | dh1 | yes | GdCl3 | |||
| 2 | 74 | |||||||||
| 5 | 75 | |||||||||
| 7 | 1 | 76 | bc1 | cry1 | dh1 | yes | GdCl3 | |||
| 9 | 2 | 77 | bc1 | cry1 | dh1 | yes | GdCl3 | |||
| 3 | 78 | |||||||||
| 02/05/2014 | cm4982-2 | 767 | data_0767_2 | 2 | 79 | bc1 | cry1 | dh1 | no | KPtCl4 |
| 3 | 80 | |||||||||
| 4 | 81 | |||||||||
| 9 | 82 | |||||||||
| 10 | 83 | |||||||||
| 13 | 84 | |||||||||
| 15 | 85 | |||||||||
| data_0767_7 | 1 | 86 | bc1 | cry1 | dh1 | no | KPtCl4 | |||
| 2 | 87 | |||||||||
| 3 | 88 | |||||||||
| 4 | 89 | |||||||||
| 5 | 90 | |||||||||
| 6 | 91 | |||||||||
| 7 | 92 | |||||||||
| 8 | 93 | |||||||||
| 9 | 94 | |||||||||
| 10 | 95 | |||||||||
| 11 | 96 | |||||||||
| 12 | 97 | |||||||||
| 14 | 98 | |||||||||
| 15 | 99 | |||||||||
| 16 | 100 | |||||||||
| data_0767_9 | 1 | 101 | bc1 | cry1 | dh1 | no | KPtCl4 | |||
| 3 | 102 | |||||||||
| 4 | 103 | |||||||||
| 5 | 104 | |||||||||
| 6 | 105 | |||||||||
| data_0767_10 | 1 | 106 | bc1 | cry1 | dh1 | no | KPtCl4 | |||
| 2 | 107 | |||||||||
| 3 | 108 | |||||||||
| 4 | 109 | |||||||||
| 5 | 110 | |||||||||
| 6 | 111 | |||||||||
| 7 | 112 | |||||||||
| 11 | 113 | |||||||||
| data_0767_11 | 1 | 114 | bc1 | cry1 | dh1 | no | KPtCl4 | |||
| 2 | 115 | |||||||||
| 3 | 116 | |||||||||
| 4 | 117 | |||||||||
| 5 | 118 | |||||||||
| 7 | 119 | |||||||||
| 8 | 120 | |||||||||
| 9 | 121 | |||||||||
| 10 | 122 | |||||||||
| 11 | 123 | |||||||||
| data_0767_13 | 2 | 124 | bc1 | cry1 | dh1 | no | KPtCl4 | |||
| 3 | 125 | |||||||||
| 6 | 126 | |||||||||
| data_0767_14 | 1 | 127 | bc1 | cry1 | dh1 | no | KPtCl4 | |||
| 2 | 128 | |||||||||
| 3 | 129 | |||||||||
| 4 | 130 | |||||||||
| data_0767_15 | 1 | 131 | bc1 | cry1 | dh1 | no | KPtCl4 | |||
| 2 | 132 | |||||||||
| 3 | 133 | |||||||||
| 4 | 134 | |||||||||
| 754 | 1 | 1_1 | 135 | bc1 | cry3 | dh2 | no | KPtCl4 | ||
| 1_2 | 136 | |||||||||
| 1_3 | 137 | |||||||||
| 1_4 | 138 | |||||||||
| 1_5 | 139 | |||||||||
| 1_6 | 140 | |||||||||
| 1_7 | 141 | |||||||||
| 1_8 | 142 | |||||||||
| 1_9 | 143 | |||||||||
| 1_10 | 144 | |||||||||
| 4 | 4_1 | 145 | bc1 | cry3 | dh2 | no | KPtCl4 | |||
| 4_2 | 146 | |||||||||
| 4_3 | 147 | |||||||||
| 4_4 | 148 | |||||||||
| 4_5 | 149 | |||||||||
| 4_6 | 150 | |||||||||
| 4_7 | 151 | |||||||||
| 4_8 | 152 | |||||||||
| 4_9 | 153 | |||||||||
| 4_10 | 154 | |||||||||
| 4_11 | 155 | |||||||||
| 5 | 5_1 | 156 | bc1 | cry3 | dh2 | no | KPtCl4 | |||
| 5_2 | 157 | |||||||||
| 5_3 | 158 | |||||||||
| 758 | 02 | 2_2 | 159 | bc3 | cry1 | dh1 | no | Os | ||
| 2_3 | 160 | |||||||||
| 03 | 3_1 | 161 | bc3 | cry1 | dh1 | no | Os | |||
| 04 | 4_2 | 162 | bc3 | cry1 | dh1 | no | Os | |||
| 4_4 | 163 | |||||||||
| 4_5 | 164 | |||||||||
| 05 | 5_1 | 165 | bc3 | cry1 | dh1 | no | Os | |||
| 5_2 | 166 | |||||||||
| 5_3 | 167 | |||||||||
| 5_4 | 168 | |||||||||
| 5_5 | 169 | |||||||||
| 06 | 6_1 | 170 | bc3 | cry1 | dh1 | no | Os | |||
| 6_2 | 171 | |||||||||
| 6_3 | 172 | |||||||||
| 6_4 | 173 | |||||||||
| 6_5 | 174 | |||||||||
| 08 | 8_1 | 175 | bc3 | cry1 | dh1 | no | Os | |||
| 10 | 10_1 | 176 | bc3 | cry1 | dh1 | no | Os | |||
| 10_2 | 177 | |||||||||
| 11 | 11_1 | 178 | bc3 | cry1 | dh1 | no | Os | |||
| 11_2 | 179 | |||||||||
| 13 | 13_1 | 180 | bc3 | cry1 | dh1 | no | Os | |||
| 13_2 | 181 | |||||||||
| 15 | 15_2 | 182 | bc3 | cry1 | dh1 | no | Os | |||
| 15_3 | 183 | |||||||||
| 15_4 | 184 | |||||||||
| 15_5 | 185 | |||||||||
| 15_6 | 186 | |||||||||
| 765 | 1 | 1_1 | 187 | bc3 | cry1 | dh1 | no | KPtCl4 | ||
| 1_3 | 188 | |||||||||
| 1_4 | 189 | |||||||||
| 1_5 | 190 | |||||||||
| 1_6 | 191 | |||||||||
| 1_7 | 192 | |||||||||
| 1_8 | 193 | |||||||||
| 1_9 | 194 | |||||||||
| 1_10 | 195 | |||||||||
| 1_11 | 196 | |||||||||
| 1_12 | 197 | |||||||||
| 2 | 2_3 | 198 | bc3 | cry1 | dh1 | no | KPtCl4 | |||
| 2_4 | 199 | |||||||||
| 2_5 | 200 | |||||||||
| 2_6 | 201 | |||||||||
| 2_7 | 202 | |||||||||
| 2_8 | 203 | |||||||||
| 2_9 | 204 | |||||||||
| 2_10 | 205 | |||||||||
| 2_11 | 206 | |||||||||
| 2_12 | 207 | |||||||||
| 2_13 | 208 | |||||||||
| 2_14 | 209 | |||||||||
| 5 | 5_4 | 210 | bc1 | cry1 | dh1 | no | Os | |||
| 5_6 | 211 | |||||||||
| 5_7 | 212 | |||||||||
| 5_8 | 213 | |||||||||
| 5_9 | 214 | |||||||||
| 30/06/2014 | cm4982-3 | 758 | 0758_3 | 3_1 | 215 | bc1 | cry1 | dh2 | no | KPtCl4 |
| 3_2 | 216 | |||||||||
| 3_3 | 217 | |||||||||
| 3_4 | 218 | |||||||||
| 0758_4 | 4_1 | 219 | bc3 | cry1 | dh2 | no | Os | |||
| 0758_5 | 5_1 | 220 | bc3 | cry1 | dh2 | no | Os | |||
| 5_2 | 221 | |||||||||
| 5_3 | 222 | |||||||||
| 5_4 | 223 | |||||||||
| 5_5 | 224 | |||||||||
| 5_6 | 225 | |||||||||
| 5_7 | 226 | |||||||||
| 5_8 | 227 | |||||||||
| 5_9 | 228 | |||||||||
| 5_10 | 229 | |||||||||
| 5_11 | 230 | |||||||||
| 5_12 | 231 | |||||||||
| 5_13 | 232 | |||||||||
| 5_14 | 233 | |||||||||
| 0758_6 | 6_1 | 234 | bc3 | cry1 | dh2 | no | Os | |||
| 6_2 | 235 | |||||||||
| 6_4 | 236 | |||||||||
| 6_5 | 237 | |||||||||
| 6_6 | 238 | |||||||||
| 0758_8 | 8_1 | 239 | bc3 | cry1 | dh2 | no | Os | |||
| 8_2 | 240 | |||||||||
| 8_3 | 241 | |||||||||
| 0758_9 | 9_1 | 242 | bc3 | cry1 | dh2 | no | Os | |||
| 9_2 | 243 | |||||||||
| 9_3 | 244 | |||||||||
| 9_4 | 245 | |||||||||
| 9_5 | 246 | |||||||||
| 9_6 | 247 | |||||||||
| 9_7 | 248 | |||||||||
| 0758_10 | 10_1 | 249 | bc3 | cry1 | dh2 | no | Os | |||
| 10_2 | 250 | |||||||||
| 10_3 | 251 | |||||||||
| 10_4 | 252 | |||||||||
| 10_5 | 253 | |||||||||
| 10_6 | 254 | |||||||||
| 0758_11 | 11_1 | 255 | bc3 | cry1 | dh2 | no | Os | |||
| 11_2 | 256 | |||||||||
| 11_3 | 257 | |||||||||
| 11_4 | 258 | |||||||||
| 11_5 | 259 | |||||||||
| 11_6 | 260 | |||||||||
| 0758_12 | 12_1 | 261 | bc1 | cry1 | dh2 | no | Os | |||
| 12_2 | 262 | |||||||||
| 12_3 | 263 | |||||||||
| 12_4 | 264 | |||||||||
| 12_5 | 265 | |||||||||
| 12_6 | 266 | |||||||||
| 12_7 | 267 | |||||||||
| 0758_13 | line | 268 | bc1 | cry1 | dh2 | no | Os | |||
| 0758_14 | 14_1 | 269 | bc1 | cry1 | dh2 | no | Os | |||
| 14_2 | 270 | |||||||||
| 0758_15 | 15_1 | 271 | bc1 | cry1 | dh2 | no | Os |
Appendix B

Appendix C
References
- Liu, Q.; Zhang, Z.; Hendrickson, W.A. Multi-crystal anomalous diffraction for low-resolution macromolecular phasing. Acta Cryst. 2011, D67, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Giordano, R.; Leal, R.M.F.; Bourenkov, G.P.; McSweeney, S.; Popov, A.N. The application of hierarchical cluster analysis to the selection of isomorphous crystals. Acta Cryst. 2012, D68, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Foadi, J.; Aller, P.; Alguel, Y.; Cameron, A.; Axford, D.; Owen, R.L.; Armour, W.; Waterman, D.G.; Iwata, S.; Evans, G. Clustering procedures for the optimal selection of data sets from multiple crystals in macromolecular crystallography. Acta Cryst. 2013, D69, 1617–1632. [Google Scholar] [CrossRef] [PubMed]
- Barends, T.R.M.; Foucar, L.; Shoeman, R.L.; Bari, S.; Epp, S.W.; Hartmann, R.; Hauser, G.; Huth, M.; Kieser, C.; Lomb, L.; et al. Anomalous signal from S atoms in protein crystallographic data from an X-ray free-electron laser. Acta Cryst. 2013, D69, 838–842. [Google Scholar] [CrossRef] [PubMed]
- White, T.A.; Barty, A.; Stellato, F.; Holton, J.M.; Kirian, R.A.; Zatsepin, N.A.; Chapman, H.N. Crystallographic data processing for free-electron laser sources. Acta Cryst. 2013, D69, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- El Omari, K.; Iourin, O.; Kadlec, J.; Fearn, R.; Hall, D.R.; Harlos, K.; Grimes, J.M.; Stuart, D.I. Pushing the limits of sulfur SAD phasing: de novo structure solution of the N-terminal domain of the ectodomain of HCV E1. Acta Cryst. 2014, D70, 2197–2203. [Google Scholar]
- Liu, Q.; Guo, Y.; Chang, Y.; Cai, Z.; Assur, Z.; Mancia, F.; Greene, M.I.; Hendrickson, W.A. Multi-crystal native SAD analysis at 6 keV. Acta Cryst. 2014, D70, 2544–2557. [Google Scholar] [CrossRef] [PubMed]
- Akey, D.L.; Brown, W.C.; Konwerski, J.; Ogata, C.M.; Smith, J.L. Use of massively multiple merged data for low-resolution S-SAD phasing and refinement of flavivirus NS1. Acta Cryst. 2014, D70, 2719–2729. [Google Scholar] [CrossRef] [PubMed]
- Zander, U.; Bourenkov, G.; Popov, A.; de Sanctis, D.; Svensson, O.; McCarthy, A.; Round, E.; Gordeliy, V.; Mueller-Dieckmann, C.; Leonard, G.A. MeshAndCollect: An automated multi-crystal data-collection workflow for synchrotron macromolecular crystallography beamlines. Acta Cryst. 2015, D71, 2328–2343. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Wang, B.-C.; Weiss, M.S. Native SAD is maturing. IUCrJ 2015, 2, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Pye, V.E.; Caffrey, M. Experimental phasing for structure determination using membrane-proteincrystals grown by the lipid cubic phase method. Acta Cryst. 2015, D71, 104–122. [Google Scholar]
- Axford, D.; Foadi, J.; Hu, N.-J.; Choudhury, H.; Iwata, S.; Beis, K.; Evans, G.; Yilmaz, A. Structure determination of an integral membrane protein at room temperature from crystals in situ. Acta Cryst. 2015, D71, 1228–1237. [Google Scholar]
- Schlichting, I. Serial femtosecond crystallography: The first five years. IUCrJ 2015, 2, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Olieric, V.; Weinert, T.; Finke, D.; Anders, C.; Li, D.; Olieric, N.; Borca, C.; Steinmetz, M.; Caffrey, M.; Jinek, M.; et al. Data-collection strategy for challenging native SAD phasing. Acta Cryst. 2016, D72, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Akey, D.L.; Terwilliger, T.C.; Smith, J.L. Efficient merging of data from multiple samples for determination of anomalous substructures. Acta Cryst. 2016, D72, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Dahmane, T.; Zhang, Z.; Assur, Z.; Brasch, J.; Shapiro, L.; Mancia, F.; Hendrickson, W.A. Structures from anomalous diffraction of native biological macromolecules. Science 2012, 336, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.; Olieric, V.; Waltersperger, S.; Panepucci, E.; Chen, L.; Zhang, H.; Zhou, D.; Rose, J.; Ebihara, A.; Kuramitsu, S.; et al. Fast native S-SAD phasing for routine macromolecular structure determination. Nat. Methods 2015, 12, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Krojer, T.; Talon, R.; Pearche, N.; Collins, P.; Douangamath, A.; Brandao-Neto, J.; Dias, A.; Marsden, B.; von Delft, F. The XChemExplorer graphical workflow tool for routine or large-scale protein-ligand structure determination. Acta Cryst. 2017, D73, 267–278. [Google Scholar]
- Pearce, N.; Krojer, T.; Bradley, A.; Collins, P.; Nowak, R.; Talon, R.; Marsden, B.; Kelm, S.; Shi, J.; Deane, C.M.; et al. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 2017, 8, 15123. [Google Scholar] [CrossRef] [PubMed]
- Roessler, C.; Kuczewski, A.; Stearns, R.; Ellson, R.; Olechno, J.; Orville, A.; Allaire, M.; Soares, A.S.; Heroux, A. Acoustic methods for high-throughput protein crystal mounting at next-generation macromolecular crystallographic beamlines. J. Synchrotron Rad. 2013, 20, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Axford, D.; Owen, R.; Aishima, J.; Foadi, J.; Morgan, A.; Robinson, J.; Nettleship, J.; Owens, R.; Moraes, I.; Fry, E.; et al. In situ macromolecular crystallography using microbeams. Acta Cryst. 2012, D68, 592–600. [Google Scholar]
- Lobley, C.M.C.; Sandy, J.; Sanchez-Weatherby, J.; Mazzorana, M.; Krojer, T.; Nowak, R.P.; Sorensen, T.L. A generic protocol for protein crystal dehydration using the HC1b humidity controller. Acta Cryst. 2016, D72, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Delageniere, S.; Brenchereau, P.; Launer, L.; Ashton, A.; Leal, R.; Veyrier, S.; Gabadinho, J.; Gordon, E.; Jones, S.; Levik, K.E.; et al. ISPyB: An information management system for synchrotron macromolecular crystallography. Bioinformatics 2011, 27, 3186–3192. [Google Scholar] [CrossRef] [PubMed]
- Welcome to VMXi. Available online: http://www.diamond.ac.uk/Beamlines/Mx/VMXi.html (accessed on 1 August 2017).
- Chapman, H.; Barty, A.; Bogan, M.; Boutet, S.; Frank, M.; Hau-Riege, P.; Marchesini, S.; Woods, B.; Bajt, S.; Benner, W.H.; et al. Femtosecond diffraction imaging with a soft-X-ray free-electron laser. Nat. Phys. 2006, 2, 839–843. [Google Scholar] [CrossRef]
- Barends, T.R.M.; Foucar, L.; Botha, S.; Doak, R.; Shoeman, R.; Nass, K.; Koglin, J.; Williams, G.; Boutet, S.; Messerschmidt, M.; et al. De novo protein crystal structure determination from X-ray free-electron laser data. Nature 2014, 505, 244. [Google Scholar] [CrossRef] [PubMed]
- Fromme, P.; Spence, J. CH. Femtosecond nanocrystallography using X-ray lasers for membrane protein structure determination. Curr. Opin. Struct. Biol. 2011, 21, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Gati, C.; Bourenkov, G.; Klinge, M.; Rehders, D.; Stellato, F.; Oberthür, D.; Yefanov, O.; Sommer, B.; Mogk, S.; Duszenko, M.; et al. Serial crystallography on in vivo grown microcrystals using synchrotron radiation. IUCrJ 2014, 1, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Stellato, F.; Oberthür, D.; Liang, M.; Bean, R.; Gati, C.; Yefanov, O.; Barty, A.; Burkhardt, A.; Fischer, P.; Galli, L.; et al. Room-temperature macromolecular serial crystallography using synchrotron radiation. IUCrJ 2014, 1, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Botha, S.; Nass, K.; Barends, T.R.M.; Kabsch, W.; Latz, B.; Dworkowski, F.; Foucar, L.; Panepucci, E.; Wang, M.; Shoeman, R.; et al. Room-temperature serial crystallography at synchrotron X-ray sources using slowly flowing free-standing high-viscosity microstreams. Acta Cryst. 2015, D71, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Coquelle, N.; Brewster, A.; Kapp, U.; Shilova, A.; Weinhausen, B.; Burghammer, M.; Colletier, J. Raster-scanning serial protein crystallography using micro- and nano-focused synchrotron beams. Acta Cryst. 2015, D71, 1184–1196. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, K.; Karplus, P.A. Better models by discarding data? Acta Cryst. 2013, D69, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Assmann, G.; Brehm, W.; Diederichs, K. Identification of rogue datasets in serial crystallography. J. Appl. Cryst. 2016, 49, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.; Roth, C.; Jo, E.; Griffith, M.; Scott, F.; Reinhart, G.; Desale, H.; Clemons, B.; Cahalan, S.; Schuerer, S.; et al. Crystal structure of a lipid G protein-coupled receptor. Science 2012, 335, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Murty, M.; Flynn, P.J. Data clustering: A review. ACM Comput. Surv. 1999, 31, 264–323. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Cryst. 2006, D62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Evans, G.; Murshudov, G. How good are my data and what is the resolution? Acta Cryst. 2013, D69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Foadi, J.; Aller, P. BLEND: Managing, Scaling and Merging Multiple Datasets. Available online: http://www.ccp4.ac.uk/tutorials/tutorial_files/blend_tutorial/BLEND_tutorial.html (accessed on 1 August 2017).
- Aller, P.; Geng, T.; Evans, G.; Foadi, J. Applications of the BLEND Software to Crystallographic Data from Membrane Proteins. In The Next Generation in Membrane Protein Structure Determination; Springer: Berlin, Germany, 2016; pp. 119–135. [Google Scholar]
- Diamond Light Source. Available online: http://www.diamond.ac.uk (accessed on 1 August 2017).
- Olson, E.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Posern, G.; Treisman, R. Actin’ together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol. 2006, 16, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Tan, S.; Richmond, T. Structure of serum response factor core bound to DNA. Nature 2002, 376, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Hassler, M.; Richmond, T. The B-box dominates SAP-1-SRFinteractions in the structure of the ternary complex. J. EMBO 2001, 20, 3018–3028. [Google Scholar] [CrossRef] [PubMed]
- Zaromytidou, A.; Miralles, F.; Treisman, R. MAL ternary complex factor use different mechanisms to contact a common surface on the serum response factor DNA-binding domain. Mol. Cell Biol. 2006, 26, 4134–4148. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A language and environment for statistical computing. Available online: https://www.R-project.org (accessed on 1 August 2017).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004, D60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997, D53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Douangamath, A.; Aller, P.; Sanchez-Wheatherby, J.; Moraes, I.; Brandao-Neto, J. Using high-throughput in situ plate screening to evaluate the effect of dehydration on protein crystals. Acta Cryst. 2013, D69, 920–923. [Google Scholar]
- Cowtant, K. CSYMMATCH. Available online: http://www.ccp4.ac.uk/html/csymmatch.html (accessed on 1 August 2017).
- Dodson, E. CCP4 Bullettin Board. Available online: https://www.jiscmail.ac.uk/cgi-bin/webadmin?A2=ind1707&L=ccp4bb&O=A&P=7301 (accessed on 1 August 2017).







| BC | CC | DH | CO | HA | NC | SN |
|---|---|---|---|---|---|---|
| bc1 | cry2 | no | no | no | 13 | 1 |
| bc1 | cry1 | no | no | no | 14 | 2 |
| bc2 | cry1 | no | no | no | 5 | 3 |
| bc1 | cry1 | dh1 | no | no | 7 | 4 |
| bc1 | cry1 | dh1 | no | KlCl6 | 6 | 5 |
| bc1 | cry1 | dh1 | no | Tantalum | 1 | 6 |
| bc1 | cry1 | dh1 | no | Hg(Thi) | 3 | 7 |
| bc1 | cry1 | dh1 | no | Pt(PIP) | 1 | 8 |
| bc1 | cry1 | dh1 | yes | Pt(PIP) | 1 | 9 |
| bc1 | cry1 | dh1 | no | KAu(CN)2 | 3 | 10 |
| bc1 | cry1 | dh1 | yes | KAu(CN)2 | 1 | 11 |
| bc1 | cry1 | dh1 | no | Hg(Ace) | 1 | 12 |
| bc1 | cry1 | dh1 | no | K2PtCl4 | 59 | 13 |
| bc3 | cry1 | dh1 | no | K2PtCl4 | 23 | 14 |
| bc1 | cry1 | dh2 | no | K2PtCl4 | 4 | 15 |
| bc1 | cry3 | dh2 | no | K2PtCl4 | 24 | 16 |
| bc1 | cry1 | dh1 | yes | K2PtCl4 | 4 | 17 |
| bc1 | cry1 | dh1 | no | Hg(PMA) | 1 | 18 |
| bc1 | cry1 | dh1 | no | K2PtI6 | 1 | 19 |
| bc1 | cry1 | dh1 | yes | OsCl3 | 1 | 20 |
| bc1 | cry1 | dh1 | yes | AgN | 1 | 21 |
| bc1 | cry1 | dh1 | yes | I3C(magic triangle) | 1 | 22 |
| bc1 | cry1 | dh1 | yes | GdCl3 | 9 | 23 |
| bc1 | cry1 | dh1 | no | Os | 5 | 24 |
| bc3 | cry1 | dh1 | no | Os | 28 | 25 |
| bc1 | cry1 | dh2 | no | Os | 11 | 26 |
| bc3 | cry1 | dh2 | no | Os | 42 | 27 |
| Dataset Number | Rmeas | Rpim | Completeness (%) | Multi-Plicity | Resolution CC1/2 | Resolution Mn(I/sd) | Resolution Max |
|---|---|---|---|---|---|---|---|
| 1 | 0.472 | 0.217 | 93.5 | 3.8 | 4.39 | 5.82 | 4.00 |
| 2 | 0.537 | 0.298 | 92.3 | 2.7 | 4.97 | 5.70 | 4.00 |
| 3 | 2.107 | 1.430 | 97.9 | 2.6 | 4.00 | 4.00 | 4.00 |
| 4 | 0.328 | 0.137 | 99.9 | 6.5 | 5.85 | 5.93 | 4.00 |
| 5 | 0.532 | 0.311 | 78.7 | 2.8 | 6.21 | 6.57 | 4.00 |
| 6 | 1.510 | 0.788 | 71.5 | 3.5 | 5.77 | 6.18 | 4.00 |
| 7 | 0.212 | 0.104 | 99.9 | 6.4 | 4.08 | 4.37 | 4.00 |
| 7 final dataset | 0.277 | 0.112 | 98.9 | 4.4 | 3.80 | 4.39 | 3.80 |
| Cluster Number | Rmeas | Rpim | Completeness (%) | Multi-Plicity | Resolution (CC1/2 = 0.3) | Resolution (Mn(I/sd) = 2) | Resolution Max |
|---|---|---|---|---|---|---|---|
| 14 | 0.979 | 0.398 | 99.9 | 6.2 | 4.91 | 5.04 | 4.00 |
| 16 | 0.958 | 0.260 | 99.8 | 13.7 | 4.24 | 4.36 | 4.00 |
| 17 | 1.758 | 0.437 | 99.7 | 16.1 | 4.27 | 4.35 | 4.00 |
| 9 | 0.778 | 0.377 | 99.5 | 4.2 | 4.92 | 5.62 | 4.00 |
| 13 | 0.618 | 0.210 | 99.2 | 7.6 | 4.00 | 4.37 | 4.00 |
| 11 | 0.632 | 0.290 | 97.9 | 4.3 | 5.20 | 4.70 | 4.00 |
| 10 | 0.707 | 0.377 | 97.2 | 3.2 | 5.41 | 5.11 | 4.00 |
| 7 | 0.337 | 0.158 | 92.2 | 3.7 | 4.00 | 4.58 | 4.00 |
| Cluster Number | Datasets Filtered | Rmeas | Rpim | Completeness (%) | Multi-Plicity | Resolution CC1/2 | Resolution Mn(I/sd) | Resolution Max |
|---|---|---|---|---|---|---|---|---|
| 4 | 6 | 1.542 | 0.544 | 99.3 | 8.5 | 4.25 | 4.86 | 3.00 |
| 6 | none | 17.651 | 7.004 | 98.6 | 7.0 | 5.13 | 6.26 | 3.00 |
| 8 | 7 | 9.365 | 3.128 | 99.4 | 9.8 | 5.68 | 5.01 | 3.00 |
| 9 | 4,7 | 9.365 | 3.128 | 99.4 | 9.8 | 5.68 | 5.01 | 3.00 |
| 10 | 1,4,6,7,10 | 0.733 | 0.223 | 99.4 | 10.5 | 3.98 | 4.59 | 3.00 |
| Cluster Number | Datasets Filtered | Rmeas | Rpim | Completeness (%) | Multi-Plicity | Resolution CC1/2 | Resolution Mn(I/sd) | Resolution Max |
|---|---|---|---|---|---|---|---|---|
| 4 | 6 | 0.366 | 0.158 | 99.0 | 5.4 | 4.39 | 4.98 | 3.50 |
| 8 | 7 | 2.875 | 0.927 | 99.5% | 9.9 | 5.17 | 4.98 | 3.50 |
| 6 | none | 1.876 | 0.691 | 99.3 | 7.4 | 5.04 | 5.76 | 3.50 |
| 9 | 4,7 | 2.875 | 0.927 | 99.5 | 9.9 | 5.17 | 4.98 | 3.50 |
| 10 | 1,4,6,7,10 | 0.383 | 0.115 | 99.8 | 11.0 | 4.01 | 4.69 | 3.50 |
| Cluster Number | Datasets Filtered | Rmeas | Rpim | Completeness (%) | Multi-Plicity | Resolution CC1/2 | Resolution Mn(I/sd) | Resolution Max |
|---|---|---|---|---|---|---|---|---|
| 4 | 6 | 0.328 | 0.148 | 97.5 | 4.8 | 4.37 | 5.01 | 3.50 |
| 8 | 7 | 0.704 | 0.256 | 98.5 | 7.5 | 5.17 | 4.98 | 3.50 |
| 6 | none | 1.314 | 0.504 | 99.5 | 7.0 | 5.31 | 5.84 | 3.50 |
| 9 | 4,7 | 0.704 | 0.256 | 98.5 | 7.5 | 6.07 | 4.96 | 3.50 |
| 10 | 1,4,6,7,10 | 0.383 | 0.115 | 99.8 | 11.0 | 4.01 | 4.69 | 3.50 |
| Dataset | Resolution Low (Å) | Resolution High (Å) | Completeness (%) | Rwork | Rfree |
|---|---|---|---|---|---|
| serial25_01.mtz | 98.00 | 3.80 | 91.87 | 0.36 | 0.42 |
| serial02_01.mtz | 104.49 | 3.50 | 89.71 | 0.41 | 0.51 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mylona, A.; Carr, S.; Aller, P.; Moraes, I.; Treisman, R.; Evans, G.; Foadi, J. A Novel Approach to Data Collection for Difficult Structures: Data Management for Large Numbers of Crystals with the BLEND Software. Crystals 2017, 7, 242. https://doi.org/10.3390/cryst7080242
Mylona A, Carr S, Aller P, Moraes I, Treisman R, Evans G, Foadi J. A Novel Approach to Data Collection for Difficult Structures: Data Management for Large Numbers of Crystals with the BLEND Software. Crystals. 2017; 7(8):242. https://doi.org/10.3390/cryst7080242
Chicago/Turabian StyleMylona, Anastasia, Stephen Carr, Pierre Aller, Isabel Moraes, Richard Treisman, Gwyndaf Evans, and James Foadi. 2017. "A Novel Approach to Data Collection for Difficult Structures: Data Management for Large Numbers of Crystals with the BLEND Software" Crystals 7, no. 8: 242. https://doi.org/10.3390/cryst7080242
APA StyleMylona, A., Carr, S., Aller, P., Moraes, I., Treisman, R., Evans, G., & Foadi, J. (2017). A Novel Approach to Data Collection for Difficult Structures: Data Management for Large Numbers of Crystals with the BLEND Software. Crystals, 7(8), 242. https://doi.org/10.3390/cryst7080242