From Initial Hit to Crystal Optimization with Microseeding of Human Carbonic Anhydrase IX—A Case Study for Neutron Protein Crystallography


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Crystallization
2.3. Deuterium Labeling and X-ray Analysis
3. Results & Discussion
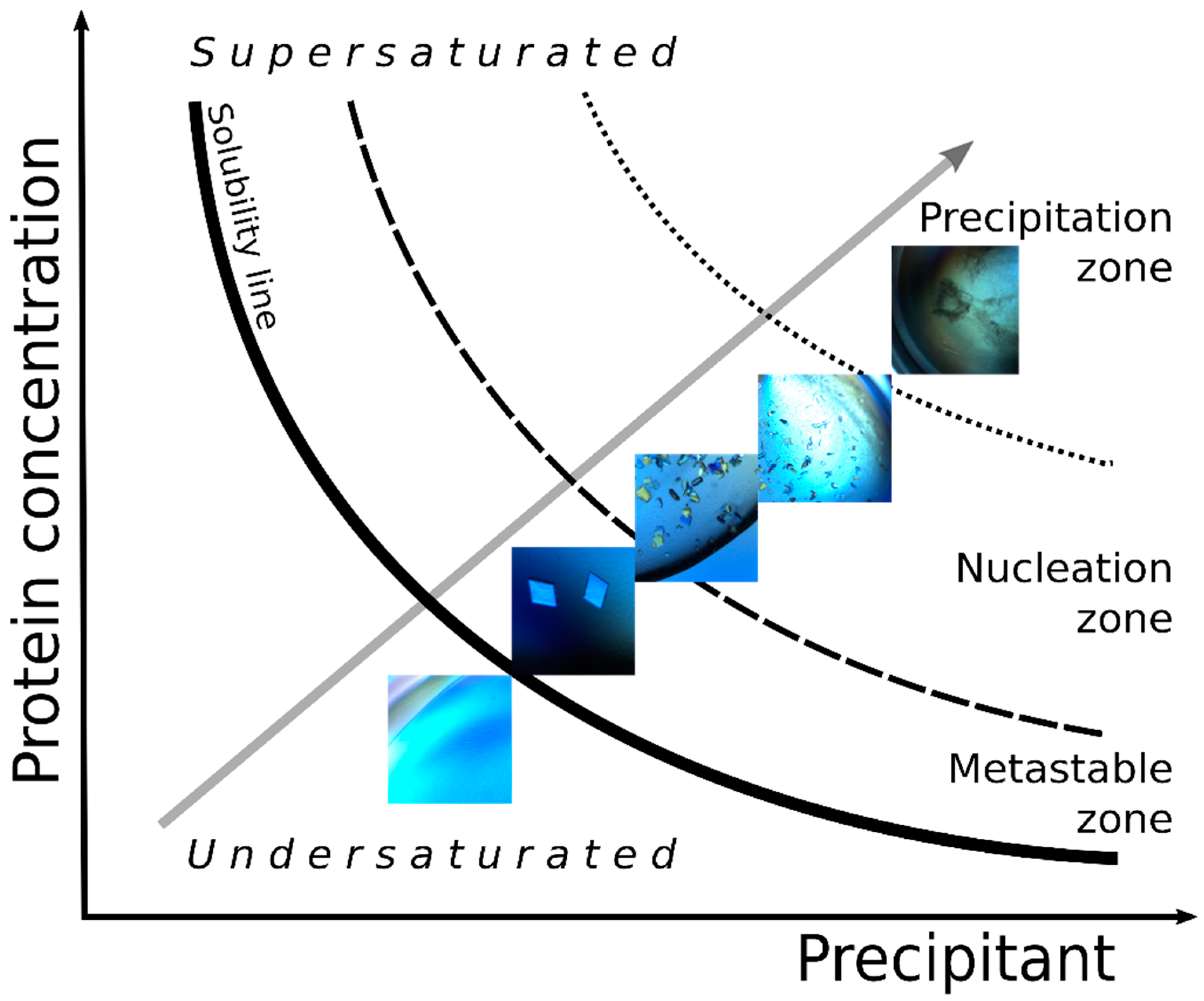
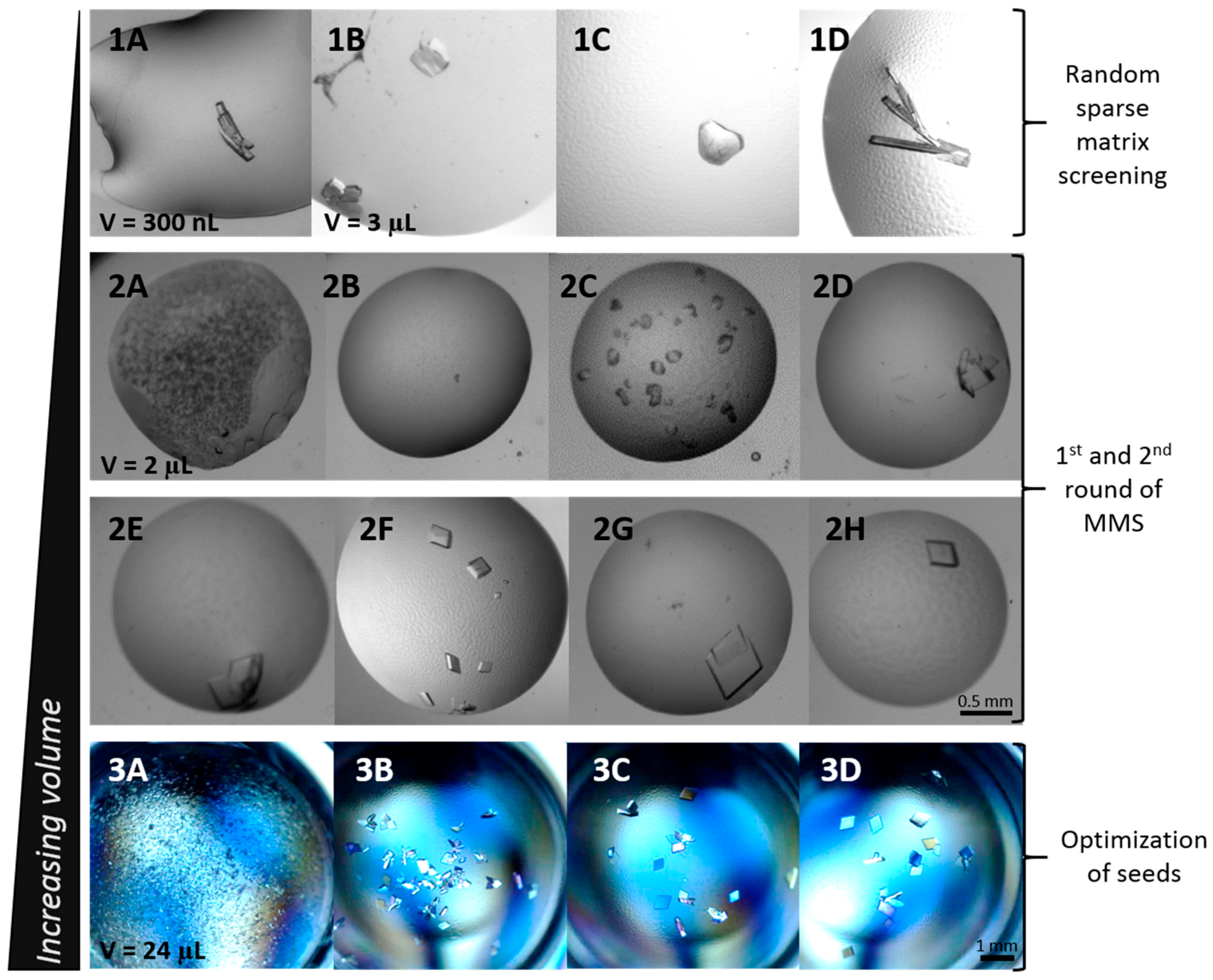
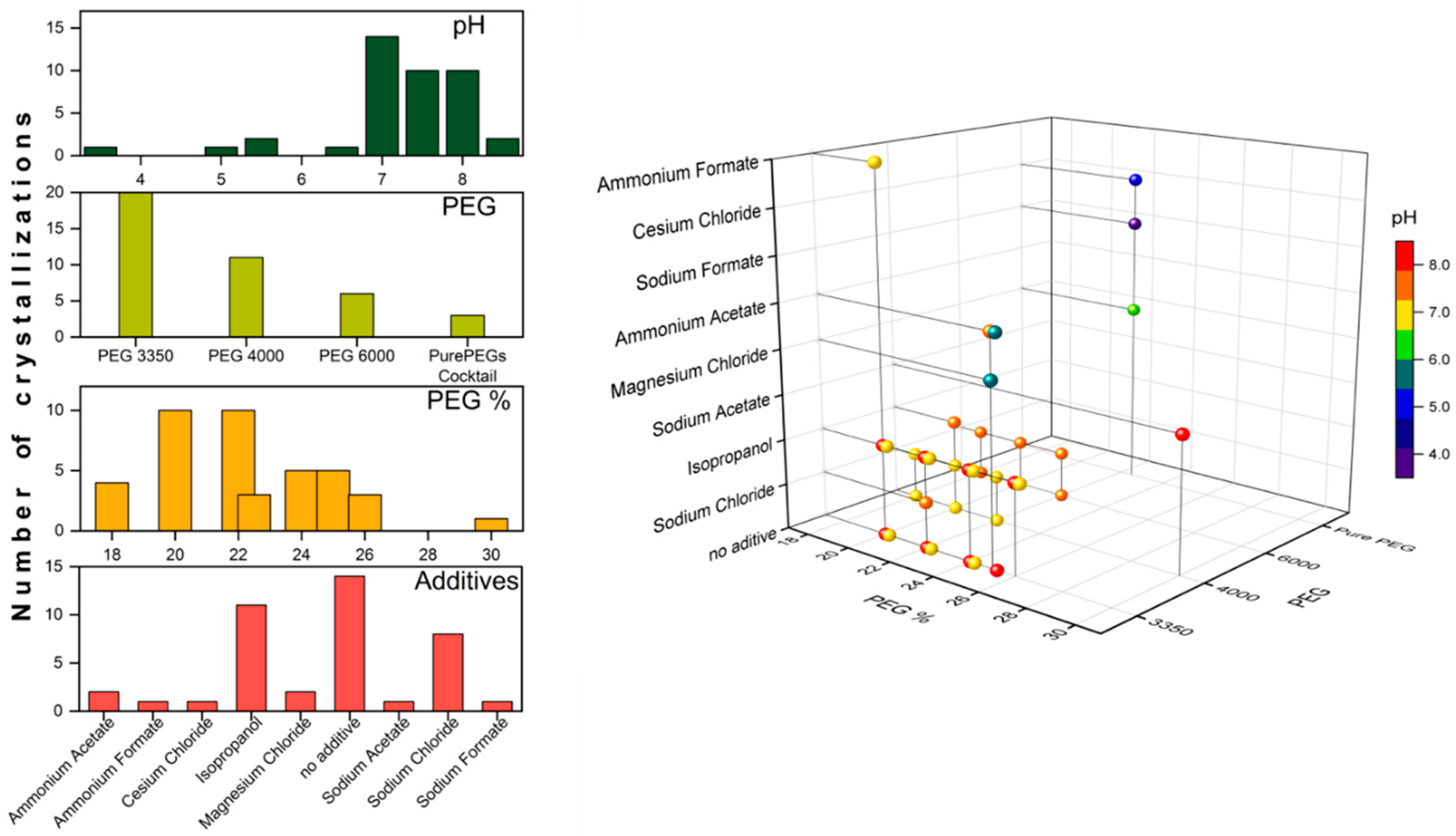
3.1. Initial Screening
3.2. Microseed Matrix Screening (MMS)
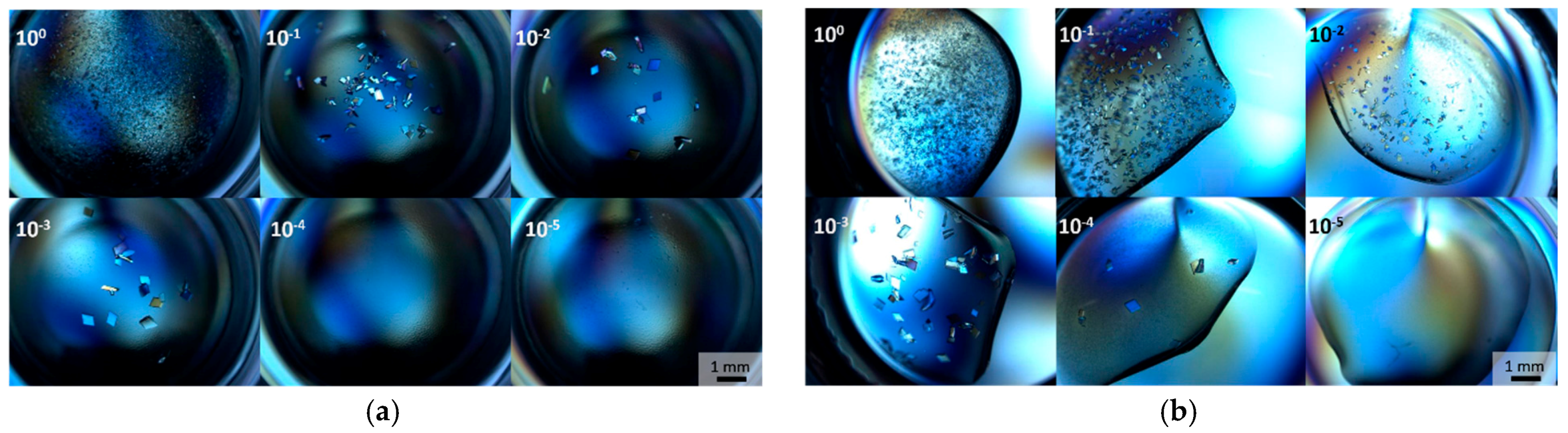
3.3. Optimization with Seed Concentration
3.4. Deuterated CA IX SV and Large Volume Crystallization
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ng, J.D.; Gavira, J.A.; Garcı́a-Ruı́z, J.M. Protein crystallization by capillary counterdiffusion for applied crystallographic structure determination. J. Struct. Biol. 2003, 142, 218–231. [Google Scholar] [CrossRef]
- McPherson, A.; Gavira, J.A. Introduction to protein crystallization. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2014, 70, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Blakeley, M.P.; Hasnain, S.S.; Antonyuk, S.V. Sub-atomic resolution X-ray crystallography and neutron crystallography: Promise, challenges and potential. IUCrJ 2015, 2, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Meilleur, F.; Coates, L.; Cuneo, M.; Kovalevsky, A.; Myles, D. The neutron macromolecular crystallography instruments at Oak Ridge National Laboratory: Advances, challenges, and opportunities. Crystals 2018, 8, 388. [Google Scholar] [CrossRef]
- O’Dell, W.B.; Swartz, P.D.; Weiss, K.L.; Meilleur, F. Crystallization of a fungal lytic polysaccharide monooxygenase expressed from glycoengineered Pichia pastoris for X-ray and neutron diffraction. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2017, 73, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Ohlin, M.; von Schantz, L.; Schrader, T.E.; Ostermann, A.; Logan, D.T.; Fisher, S.Z. Crystallization, neutron data collection, initial structure refinement and analysis of a xyloglucan heptamer bound to an engineered carbohydrate-binding module from xylanase. Acta Crystallogr. F Struct. Biol. Cryst. 2015, 71, 1072–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzoni, F.; Saraboji, K.; Sprenger, J.; Kumar, R.; Noresson, A.-L.; Nilsson, U.J.; Leffler, H.; Fisher, S.Z.; Schrader, T.E.; Ostermann, A.; et al. Perdeuteration, crystallization, data collection and comparison of five neutron diffraction data sets of complexes of human galectin-3C. Acta Crystallogr. D Struct. Biol. 2016, 72, 1194–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlits, O.O.; Coates, L.; Woods, R.J.; Kovalevsky, A. Mannobiose binding induces changes in hydrogen bonding and protonation states of acidic residues in Concanavalin A as revealed by neutron crystallography. Biochemistry 2017, 56, 4747–4750. [Google Scholar] [CrossRef] [PubMed]
- Abuhammad, A.; McDonough, M.A.; Brem, J.; Makena, A.; Johnson, S.; Schofield, C.J.; Garman, E.F. “To Cross-Seed or Not To Cross-Seed”: A Pilot Study Using Metallo-β-lactamases. Cryst. Growth Des. 2017, 17, 913–924. [Google Scholar] [CrossRef]
- Obmolova, G.; Malia, T.J.; Teplyakov, A.; Sweet, R.W.; Gilliland, G.L. Protein crystallization with microseed matrix screening: Application to human germline antibody Fabs. Acta Crystallogr. F Struct. Biol. Cryst. 2014, 70, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, A.; Mac Sweeney, A.; Haber, A. Using natural seeding material to generate nucleation in protein crystallization experiments. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Blakeley, M.P.; Kalb, A.J.; Helliwell, J.R.; Myles, D.A. The 15-K neutron structure of saccharide-free Concanavalin A. Proc. Natl. Acad. Sci. USA 2004, 101, 16405–16410. [Google Scholar] [CrossRef] [PubMed]
- Gavira, J.A.; Hernandez-Hernandez, M.A.; Gonzalez-Ramirez, L.A.; Briggs, R.A.; Kolek, S.A.; Shaw Stewart, P.D. combining counter-diffusion and microseeding to increase the success rate in protein crystallization. Cryst. Growth Des. 2011, 11, 2122–2126. [Google Scholar] [CrossRef]
- Asherie, N. Protein crystallization and phase diagrams. Methods 2004, 34, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Luft, J.R.; DeTitta, G.T. A method to produce microseed stock for use in the crystallization of biological macromolecules. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 988–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, E.; Chen, J.C.; Fisher, S.Z. Neutron crystallography for the study of hydrogen bonds in macromolecules. Molecules 2017, 22, 596. [Google Scholar] [CrossRef] [PubMed]
- Fisher, Z.; Jackson, A.; Kovalevsky, A.; Oksanen, E.; Wacklin, H. Biological structures. In Experimental Methods in the Physical Sciences; Felix, F.-A., David, L.P., Eds.; Elsevier: San Diego, CA, USA, 2017; Volume 49, pp. 1–75. [Google Scholar]
- Blum, M.-M.; Tomanicek, S.J.; John, H.; Hanson, B.L.; Rüterjans, H.; Schoenborn, B.P.; Langan, P.; Chen, J.C.-H. X-ray structure of perdeuterated diisopropyl fluorophosphatase (DFPase): Perdeuteration of proteins for neutron diffraction. Acta Crystallogr. F Struct. Biol. Cryst. 2010, 66, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, I.; Kusaka, K.; Hosoya, T.; Niimura, N.; Ohhara, T.; Kurihara, K.; Yamada, T.; Ohnishi, Y.; Tomoyori, K.; Yokoyama, T. Neutron structure analysis using the IBARAKI biological crystal diffractometer (iBIX) at J-PARC. Acta Crystallogr. D Struct. Biol. 2010, 66, 1194–1197. [Google Scholar] [CrossRef] [PubMed]
- Pastorek, J.; Pastorekova, S. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: From biology to clinical use. Semin. Cancer Biol. 2015, 31, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazokaite, J.; Niemans, R.; Dudutiene, V.; Becker, H.M.; Leitans, J.; Zubriene, A.; Baranauskiene, L.; Gondi, G.; Zeidler, R.; Matuliene, J.; et al. Novel fluorinated carbonic anhydrase IX inhibitors reduce hypoxia-induced acidification and clonogenic survival of cancer cells. Oncotarget 2018, 9, 26800–26816. [Google Scholar] [CrossRef] [PubMed]
- Mahon, B.P.; Bhatt, A.; Socorro, L.; Driscoll, J.M.; Okoh, C.; Lomelino, C.L.; Mboge, M.Y.; Kurian, J.J.; Tu, C.; Agbandje-McKenna, M.; et al. The Structure of Carbonic Anhydrase IX Is Adapted for Low-pH Catalysis. Biochemistry 2016, 55, 4642–4653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ireton, G.C.; Stoddard, B.L. Microseed matrix screeningto improve crystals of yeast cytosine deaminase. Acta Crystallogr. D Struct. Biol. 2004, 60, 801. [Google Scholar] [CrossRef]
- Koruza, K.; Lafumat, B.; Végvári, Á.; Knecht, W.; Fisher, S.Z. Deuteration of human carbonic anhydrase for neutron crystallography: Cell culture media, protein thermostability, and crystallization behavior. Arch. Biochem. Biophys. 2018, 645, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Budayova-Spano, M.; Fisher, S.Z.; Dauvergne, M.-T.; Agbandje-McKenna, M.; Silverman, D.N.; Myles, D.A.A.; McKenna, R. Production and X-ray crystallographic analysis of fully deuterated human carbonic anhydrase II. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2006, 62, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, L.; Moulin, M.; Haertlein, M.; Meilleur, F.; Christianson, D.W. Expression, purification, assay, and crystal structure of perdeuterated human arginase I. Arch. Biochem. Biophys. 2007, 465, 82–89. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Precipitant | Buffer | pH | Additive/Extra | Crystallization Set-Up | Correlation to Figure 2 |
|---|---|---|---|---|---|
| 20% PEG 3350 | None | n/a | 0.2 M Ammonium Formate | Hanging drop | 1A 1 |
| 20–24% PEG 3350 | 0.1 M Tris | 7.0 | None | Sitting drop | n/a |
| 25% PEG 3350 | 0.1 M HEPES | 7.5 | None | Sitting drop | 2H |
| 20–24% PEG 3350 | 0.1 M Tris | 8.0 | None | Sitting drop | n/a |
| 25% PEG 3350 | 0.1 M Tris | 8.5 | None | Sitting drop | 2G |
| 25% PEG 3350 | 0.1 M Tris | 5.5 | 0.2 M Magnesium Chloride | Sitting drop | 2C |
| 20–26% PEG 3350 | 0.1 M Tris | 7.0 | 4–10% 2-propanol | Sitting drop | n/a |
| 25% PEG 3350 | 0.1 M HEPES | 7.5 | 0.2 M Ammonium Acetate | Sitting drop | 2D |
| 22% PEG 3350 | 0.1 M Tris | 7.5 | 0.2 M Sodium Chloride | Hanging drop, microbatch | 1B 1 |
| 20–26% PEG 3350 | 0.1 M Tris | 8.0 | 4–10% 2-propanol | Sitting drop | n/a |
| 18–22% PEG 4000 | 0.1 M Tris | 7.0 | None | Sitting drop | n/a |
| 18–22% PEG 4000 | 0.1 M Tris | 7.0 | 0.2 M Sodium Chloride | Hanging drop, microbatch | 1C 1 |
| 20% PEG 4000 | 0.1 M HEPES | 7.5 | 10% 2-propanol | Sitting drop | 2E |
| 22–26% PEG 4000 | 0.1 M Tris | 8.0 | 10% 2-propanol | Sitting drop | n/a |
| 30% PEG 4000 | 0.1 M Tris | 8.5 | 0.2 M Sodium Acetate | Sitting drop, microbatch | 2F, 3A–D |
| 18–22% PEG 6000 | 0.1 M Tris | 7.5 | None | Sitting drop | n/a |
| 18–22% PEG 6000 | 0.1 M Tris | 7.5 | 0.2 M Sodium Chloride | Hanging drop | 1D 1 |
| 22.5% PurePEGs Cocktail | 0.1 M Citric Acid | 3.5 | 0.3 M Sodium Formate | Sitting drop | n/a |
| 22.5% PurePEGs Cocktail | 0.1 M Ammonium Citrate | 5.0 | 0.3 M Cesium Chloride | Sitting drop | 2B |
| 22.5% PurePEGs Cocktail | 0.1 M Sodium Cacodylate | 6.5 | 0.3 M Magnesium Chloride | Sitting drop | 2A |
| 1.0 M Sodium Citrate | 0.1 M Sodium Cacodylate | 6.5 | None | Sitting drop | n/a |
| H/H | H/D | D/D | |
|---|---|---|---|
| Wavelength (Å) Beamline, source | 0.979 BM-30 (FIP), ESRF | 0.979 BioMAX, MAX IV laboratory | 0.979 BioMAX, MAX IV laboratory |
| Space group, unit cell | P21; a = 44.3, b = 65.1, c = 46.7; β = 115.1 | P21; a = 44.5, b = 65.4, c = 46.7; β = 115.1 | P21; a = 44.4, b = 65.1, c = 46.6; β = 114.7 |
| Resolution range (Å) | 50.0–1.77 (1.88–1.77) | 40.0–1.39 (1.42–1.39) | 40.0–1.49 (1.51–1.49) |
| Total No. of reflections | 63,296 (9199) | 327,912 (15,926) | 265,165 (13,184) |
| No. of unique reflections | 22,187 (3463) | 48,352 (2406) | 39,461 (1948) |
| Redundancy | 2.8 (2.6) | 6.8 (6.6) | 6.7 (6.8) |
| Completeness (%) | 95.1 (92.9) | 99.8 (99.1) | 99.9 (99.3) |
| 〈I/σ(I)〉 | 10.5 (2.2) | 19.9 (2.2) | 14.3 (2.2) |
| Rmerge † (%) | 6.3 (48.6) | 3.6 (78.1) | 6.0 (78.1) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koruza, K.; Lafumat, B.; Nyblom, M.; Knecht, W.; Fisher, Z. From Initial Hit to Crystal Optimization with Microseeding of Human Carbonic Anhydrase IX—A Case Study for Neutron Protein Crystallography. Crystals 2018, 8, 434. https://doi.org/10.3390/cryst8110434
Koruza K, Lafumat B, Nyblom M, Knecht W, Fisher Z. From Initial Hit to Crystal Optimization with Microseeding of Human Carbonic Anhydrase IX—A Case Study for Neutron Protein Crystallography. Crystals. 2018; 8(11):434. https://doi.org/10.3390/cryst8110434
Chicago/Turabian StyleKoruza, Katarina, Bénédicte Lafumat, Maria Nyblom, Wolfgang Knecht, and Zoë Fisher. 2018. "From Initial Hit to Crystal Optimization with Microseeding of Human Carbonic Anhydrase IX—A Case Study for Neutron Protein Crystallography" Crystals 8, no. 11: 434. https://doi.org/10.3390/cryst8110434
APA StyleKoruza, K., Lafumat, B., Nyblom, M., Knecht, W., & Fisher, Z. (2018). From Initial Hit to Crystal Optimization with Microseeding of Human Carbonic Anhydrase IX—A Case Study for Neutron Protein Crystallography. Crystals, 8(11), 434. https://doi.org/10.3390/cryst8110434