1. Introduction
Wolframite is an iron manganese tungstate mineral. The name is normally used to denote the family of isomorphic compounds. The crystal structure of a wolframite was first solved for MgWO
4 by Broch [
1] in 1929 with the exception of the oxygen positions and then completely determined for NiWO
4 by Keeling [
2] in 1957. The structure of wolframite, monoclinic with space group
P2/
c, is adopted by all tungstates
AWO
4 with the divalent cation
A with an ionic radius in octahedral coordination
rA < 0.9 Å. In addition, CdWO
4, both
M = In and Sc niobates
MNbO
4 and tantalates
MTaO
4, and a metastable high-pressure (HP) and high-temperature polymorph in molybdates
AMoO
4 [
3] have an isostructural crystal structure. Most wolframites, and in particular tungstates and molybdates with an
A2+ cation with a completely empty or full outer
d shell, have been extensively studied for their applications in scintillating detectors for X-ray tomography, high-energy particle physics, and dosimetry devices [
4,
5,
6]. The reason behind this is their high light yield when hit by γ-particles or X-rays despite long scintillation times of a few µs [
7]. In fact, the search for different polymorphs of the scintillating wolframites with enhanced properties, i.e., a faster scintillating response, has motivated the study of these materials under high pressure conditions. In addition, wolframites with transition metals with unfilled outer
d-shells are magnetic. In particular, hübnerite MnWO
4, which shows three different antiferromagnetic phases below 13.7 K, is a type II multiferroic material [
8,
9], exhibiting ferroelectricity induced by helical magnetic ordering.
After the pioneering work of Young and Schwartz [
3], dated from 1963, in which the synthesis of different wolframite-type molybdates was reported at ~0.6 GPa and 900 °C, only two works were published about the HP behavior of wolframites during the 1990s. A detailed structural study of the wolframite structure of MgWO
4, MnWO
4, and CdWO
4 was performed at room temperature by Macavei and Schultz [
10] up to 9.3 GPa using single crystal X-ray diffraction (XRD). A Raman spectroscopy study of CdWO
4 at up to ~40 GPa was carried out by Jayaraman et al. [
11] in which a structural phase transition of CdWO
4 was found at 20 GPa. However, during the last decade, the interest in the behavior of wolframites under high pressure has inspired many works that have contributed to our understanding of their structural [
12,
13,
14,
15,
16], vibrational [
15,
16,
17,
18,
19,
20,
21], and electronic [
14,
22] properties under compression.
In the following sections, we shall review first the main features of the behavior and trends of the structure of wolframites under high pressure and the advances on the structural determination of their HP phases. Finally, the effects of pressure on the vibrational, electronic, and optical properties shall be presented with a special emphasis on CdWO4, whose HP phase has recently been fully solved.
2. The Wolframite Structure at High Pressure
The best description of the wolframite structure at ambient pressure was formulated by Kihlborg and Gebert [
23] in 1969, unfolding the structure of the Jahn–Teller distorted CuWO
4 wolframite. The wolframite structure of an
AXO
4 compound at ambient conditions (
Figure 1a) can be described as a framework of oxygen atoms in approximately hexagonal close packing with the cations (
A and
X = W), octahedrally coordinated and occupying half of the octahedral sites. In this structure, the octahedral units of the same cations share edges forming alternating zig-zag chains and conferring the structure with a layer-like
AO
XO configuration in the [100] direction. A particular case worth mentioning is CuWO
4, not included in the list of wolframites presented in the introduction. Although, according to the ionic radius of Cu, it should also crystallize in the same structure as NiWO
4 and ZnWO
4, it does it in a distorted version of the wolframite structure. In CuWO
4, as a consequence of the Jahn–Teller effect of Cu
2+ in octahedral coordination [
23,
24], the Cu
2+ ion requires a distortion that is achieved by a shear parallel to the
b-axis along each copper plane. This has as a consequence a displacement of oxygen layers, destroying the twofold symmetry and lowering the space group from
P2/
c to
P.
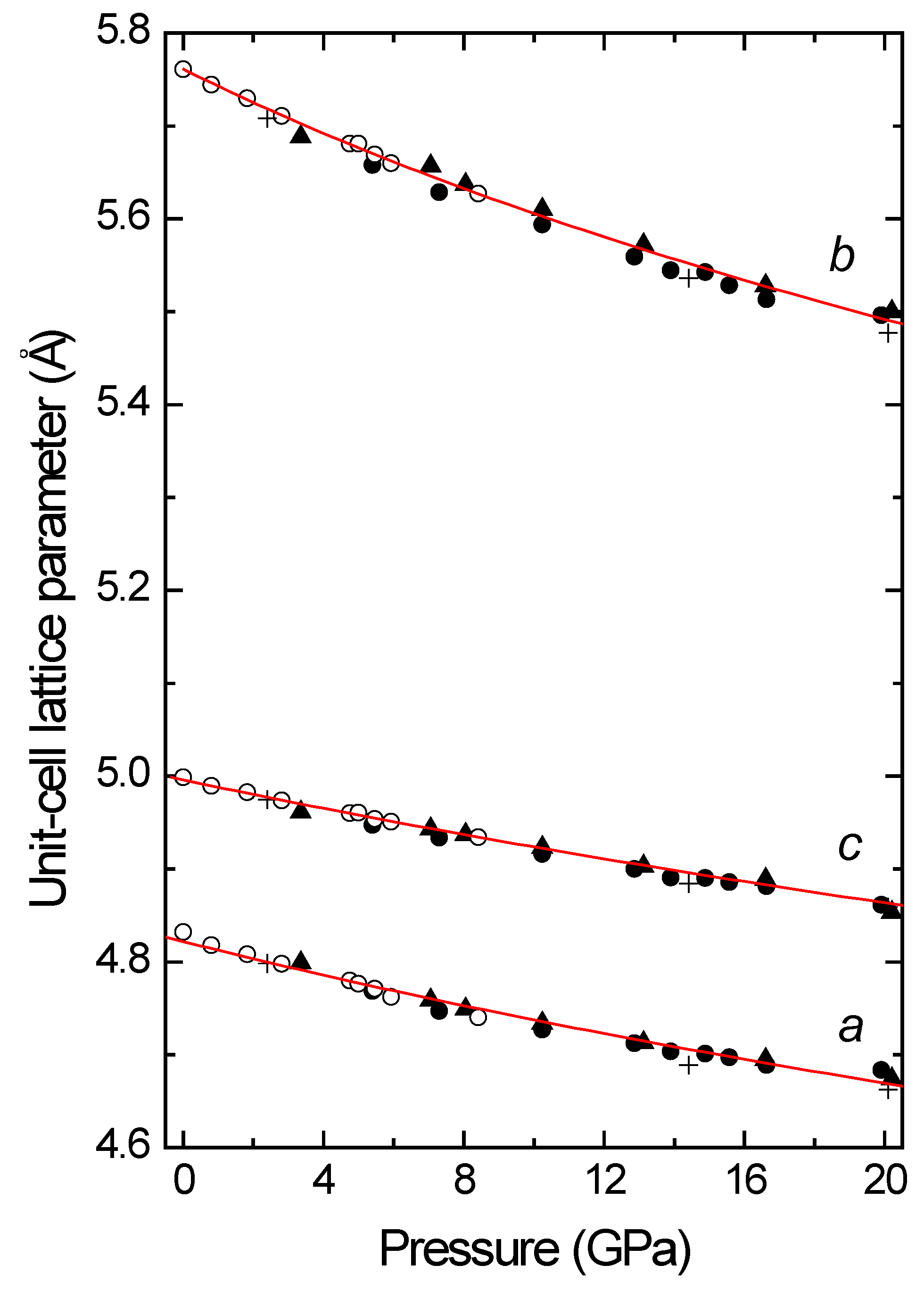
In wolframites, the monoclinic
a and
c unit-cell lattice parameters are similar, although the
c-axis is slightly larger than the
a-axis. However, the
b-axis is, in general, around 15% larger than the other axes. The monoclinic angles are always very close to 90°. As was first shown by Macavei and Shultz [
10] in MgWO
4, MnWO
4, and CdWO
4, this causes the wolframite structure to suffer an anisotropic contraction under pressure (
Figure 2). For instance, in MnWO
4 [
13], the axial compressibility (defined as
kx = (−1/x) × ∂x/∂P) of the
b-axis (
kb = 3.3(1) × 10
−3 GPa
−1) almost doubles the axis compressibility of
a and
c (
ka = 2.0(1) × 10
−3 GPa
−1 and
kc = 1.6(1) × 10
−3 GPa
−1). The same anisotropic compression has been found in CdWO
4, MgWO
4, and other wolframites [
10,
11,
12,
13,
14]. This fact, together with a continuous increase in the monoclinic β angle, indicates that, under compression, the monoclinic structure tends to distort, at least in tungstates. We shall show in the next section that this continuous distortion causes a phase transition to a structure with a lower symmetry in most wolframite-type tungstates. The only exception is CdWO
4, which increases its space-group symmetry at the phase transition. In the structure of wolframite, the octahedra of different cations only share corners between them, while octahedra of the same cation share both corners and edges. Hence, we can isolate the
AO
6 and
XO
6 polyhedral units as independent blocks in the structure. In this picture, each type of polyhedra would respond differently to the effect of pressure. In particular, one would expect the
XO
6, with high valence (6
+ in W or Mo, and 5
+ in Ta or Nb) to be much less compressible than the
A cations with 2
+ or 3
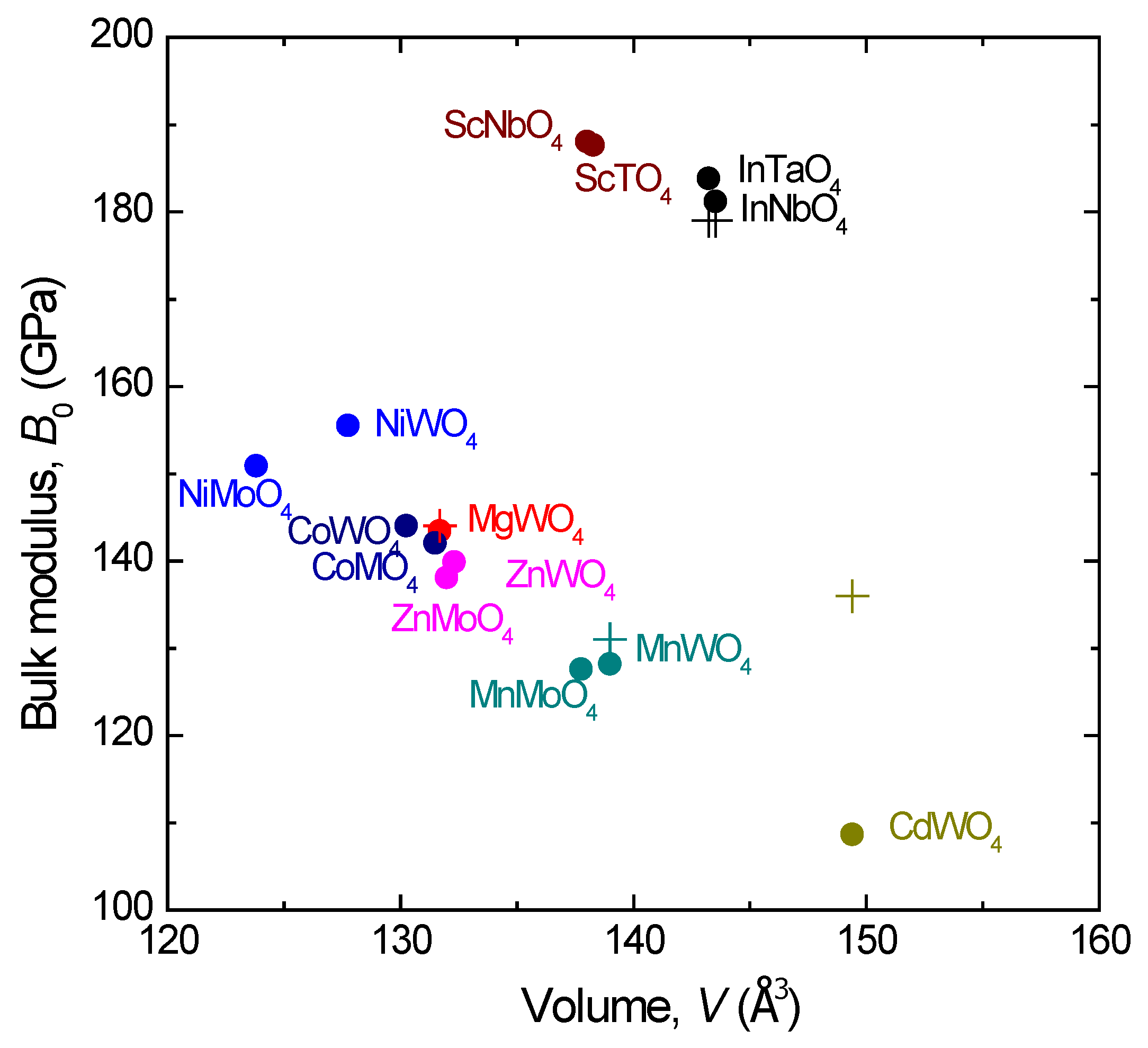
+ valences. Such an approach has often been used to study the bulk compressibility of scheelites, a structure adopted by tungstates with
A cations with a large ionic radius. However, differently from wolframites, in scheelites, the also-isolated
X cations are tetrahedrally coordinated. Therefore, the
XO
4 polyhedra are almost pressure-independent, and the bulk modulus is very well predicted by the empirical equation
B0 = N·
Z/
dA–O3, first proposed by Hazen and Finger [
25], which assumes that the bulk modulus is proportional (N being the proportion constant) to the formal charge of the
A cation,
Z, and inversely proportional to the cation–anion distance to the third power
dA–O3. In the case of scheelites, this proportion constant is N = 610 [
26]. The question is how such a model would work for wolframites. In
Figure 3, we show the estimated and experimentally available bulk modulus of all known wolframites. The agreement is excellent considering N = 661 for wolframite-type tungstates and molybdates and N = 610 for tantalates and niobates, indicating that assuming that the
XO
6 in wolframites is a rigid and almost pressure-incompressible unit is justified in terms of the bulk modulus. In
Section 4, we shall study how this approximation works in terms of describing the Grüneisen parameters of the Raman-active modes. The experimental bulk modulus values are shown in
Table 1.
Figure 3 also shows that the bulk moduli of tantalates and niobates is around 30% larger than the bulk modulus of tungstates. This fact, a result of a higher valence of the
A cation (3
+ instead of 2
+), reinforces the negligible effect of pressure on the
XO
6 polyhedra since the empirical formula is also valid despite the valence of Ta and Nb is 5
+ instead of 6
+.
Regarding the atomic positions under compression in wolframites, while the oxygen atoms barely change their positions, the cations tend to shift along the high symmetry
b-axis [
10]; the
A cations either shift down or up depending on the compound, but the W cations largely shift down along the
b-axis. For tantalates and niobates, no reliable atomic positions exist under pressure.
3. Phase Transitions
Under compression, most wolframites undergo a phase transformation to a different polymorph, with tungstates transiting at around 20 GPa, and InTaO
4 and ScNbO
4 doing so a few GPa below. However, the post-wolframite phase depends on the compound. Thus, while tungstates, except CdWO
4, apparently transform to a triclinic version of wolframites, similar to that of CuWO
4, with space group
P and a similar unit-cell, tantalates and niobates also do it but to another distorted version of wolframite that keeps the same space group
P2/
c. CuWO
4 and CdWO
4 are the only (pseudo)wolframites that, under pressure, increase their space-group symmetry, with CuWO
4 transforming to a normal wolframite structure with space group
P2/
c, despite keeping the Jahn–Teller distortion, and CdWO
4 introducing a screw axis in space group
P2
1/
c and doubling the unit cell. The solution of the HP phase of the tungstates has been approached unsuccessfully with powder X-ray diffraction in ZnWO
4 and MgWO
4 [
12] and with single crystal X-ray diffraction in MnWO
4 [
13]. However, based on the careful indexation of the observed reflections, the study of the systematic extinctions of the HP phase, and the number of active Raman modes observed in the HP phase, which we shall show in the next section, a triclinic structure is proposed as the post-wolframite structure of normal wolframites. This proposed post-wolframite structure would be very similar to the low-pressure phase, but would be described as being in space group
P. Unfortunately, in the particular case of MnWO
4, the single crystal, despite of a small volume collapse of only 1% in the phase transition, dramatically deteriorates (
Figure 4) with the appearance of more than two triclinic HP domains during the phase transition coexisting with the monoclinic low-pressure phase. This fact prevents a correct integration of the reflection intensities and therefore an accurate determination of the atomic positions in the HP phase. Since the experiments in MnWO
4 were carried out using Ne as a pressure-transmitting medium, the phase coexistence observed is likely inherent to the properties of the sample and not caused by non-hydrostatic effects. The study of this issue deserves future efforts.
In the previous section, we mentioned that CdWO
4, with Cd having an ionic radius in octahedral coordination above 0.9 Å, is just above the limit to be a wolframite at ambient pressure. This implies that the Cd–O distances are larger than the
A–O distances in the remaining wolframites and that its bulk modulus is therefore the lowest one in the series (
Figure 3). In the extreme case of MgWO
4, the Cd–O distances are around 12% longer than the Mg–O distances and the bulk modulus of MgWO
4 is 25% larger than that of CdWO
4. These differences have an influence in the phase transition of CdWO
4 that emerge for instance in the Raman spectrum of the HP phase of CdWO
4, presenting 36 instead of 18 modes observed in the post-wolframite phase of normal wolframites (
Section 4). In fact, such an increase in modes relates to the doubling of the unit cell that the post-wolframite of CdWO
4 presents above 20 GPa. The structure (
Figure 1c) was solved with single-crystal X-ray diffraction at 20 GPa [
14]. According to Macavei and Shultz [
10], the
y coordinate of Cd moves fast under pressure. In the phase transition, the
a-axis of the HP unit cell remains in the [100] direction of the low-pressure cell, increasing its length, while the new [010] and [001] directions form from the [
] and [
] directions of the low-pressure wolframite cell, respectively. Such a transformation, which can be described by the following transformation matrix [
] implies a doubling of the unit cell from
Z = 2 in the wolframite structure to the
Z = 4 of the post-wolframite of CdWO
4. Such a phase transition gives rise to the formation of a screw axis in the [010] direction of the HP phase and therefore a space-group symmetry increase from
P2/
c to
P2
1/
c. Regarding the coordination of the tungsten and cadmium ions, it is increased to 7-fold and 6+1-fold, respectively, when the phase transition occurs (
Figure 1c). This coordination increase contrasts with the phase transition undergone by normal wolframites, which keep the same octahedral coordination for both cations up to the highest pressure reached according to Raman spectroscopy in the case of the other tungstates. According to powder X-ray diffraction, a coordination increase occurs at the phase transition in tantalates and niobates as well [
15,
16]. Considering that the Cd–O distances are abnormally large in the wolframite-type structure, the coordination increase associated with the phase transition relates well with CdWO
4, which has a more compact and stable structure that differs more from the wolframite-type structure than the HP phase of the wolframite-type tungstates that keep the cationic coordination. This coordination change is directly related to a band-gap energy drop associated with the phase transition as we shall show in
Section 4 [
14].
In the case of InTaO
4 and InNbO
4, both compounds undergo phase transitions at 13 and 10.8 GPa, respectively. These are pressures around 10 GPa below the phase transition of wolframite-type tungstates. This indicates that, when the
X cation is substituted by Ta or Nb, the wolframite structure becomes less robust, probably, as the result of the lowering of the nominal charge of the ion and therefore weakening the
X–O bond. Under high pressure, InTaO
4 and InNbO
4 undergo the same phase transition to a structure (
Figure 1b) solved with powder X-ray diffraction [
15,
16]. Their post-wolframite structure consists on a packing of the wolframite-type structure with the
X cations increasing their coordination from 6 to 8-fold filling the channels that the alternating
AO
XO zigzag chains create in the [001] direction in the wolframite structure. This alternating pattern is kept in the HP phase but with the
A and
X cations having an eight-fold coordination in the HP phase. The coordination increase in both kind of cations and the packing increase generate a different interaction between the
AO
8 and
XO
8 polyhedra that now share also edges in addition to corners between them. This implies that they cannot longer be considered as separated blocks.
In order to conclude this section, we shall comment on the case of CuWO
4, which, though it presents a distorted triclinic version of the monoclinic wolframite structure at ambient pressure, also transforms to the monoclinic wolframite structure above ~9 GPa [
23]. As we explained in the previous section, the low-pressure structure of CuWO
4 is also formed by CuO
6 and WO
6 polyhedra arranged in a very similar way to wolframite. However, due to the degeneracy breaking of the 3
d9 orbitals of Cu
2+ into five electronic levels (two singlets and one doublet) as a result of the Jahn–Teller effect, two of the six Cu–O distances are longer than the other four, thus lowering its local structure from quasi
Oh to quasi
D4h symmetry. Under high pressure, the two longest Cu–O distances reduce; however, before quenching the Jahn–Teller distortion, the system finds an easy distortion direction, and the elongated axes of the CuO
6 polyhedra shift from the [
] in the low-pressure phase to the [101] in the HP phase. Thus, the structure transforms to a wolframite structure in space group
P2/
c that still accommodates the Jahn–Teller distortion of Cu
2+.
4. Raman Spectroscopy
Raman spectroscopy is a technique used to observe vibrational modes in a solid and is one of the most informative probes for studies of material properties under HP [
24]. Since the study performed two decades ago by Fomichev et al. in ZnWO
4 and CdWO
4 [
25], the Raman spectra of wolframite-type tungstates have been extensively characterized. Studies have been carried out for synthetic crystals [
17,
18,
19,
20] and minerals [
26] and have been also performed for InTaO
4 [
15] and InNbO
4 [
16]; however, the characterization of the Raman-active vibrations in wolframite-type molybdates is missing. Indeed, the Raman spectra of CoMoO
4, MnMoO
4, and MgMoO
4 have been characterized [
27,
28,
29], but for polymorphs different from wolframite.
According to group-theory analysis, a crystal structure isomorphic to wolframite has 36 vibrational modes at the Γ point of the Brillouin zone: Γ = 8A
g + 10B
g + 8A
u + 10B
u. Three of these vibrations correspond to acoustic modes (A
u + 2B
u) and the rest are optical modes, 18 of which are Raman active (8A
g + 10B
g) and 15 of which are infrared active (7A
u + 8B
u). Typical Raman spectra, taken from the literature [
17,
18,
19,
20] of wolframite-type tungstates are shown in
Figure 5. In the figure, it can be seen that the frequency distribution (and intensity) of the Raman-active modes is qualitatively similar in CdWO
4, ZnWO
4, MnWO
4, and MgWO
4. Since Raman-active vibrations correspond either to A
g or B
g modes, polarized Raman scattering and selection rules can be combined to identify the symmetry of modes [
30,
31]. The expected 18 Raman modes have been measured for CdWO
4, ZnWO
4, MnWO
4, and MgWO
4, and 15 modes for CoWO
4, FeWO
4, and NiWO
4. The frequencies of the modes are summarized in
Table 2. As we commented above, the frequency distribution of modes is similar in all tungstates, with 4 high-frequency modes, two isolated modes around 500–550 cm
−1, and the remaining 12 modes at frequencies below 450 cm
−1 in all of the compounds.
The similitude among the Raman spectra of wolframite-type tungstates can be explained, as a first approximation, by the fact that Raman modes can be classified as internal and external modes with respect to the WO
6 octahedron [
19]. Six internal stretching modes are expected to arise from the WO
6 octahedron. Four of them should have A
g symmetry and the other two B
g symmetry. Since the W atom is heavier than any of the divalent cations in
AWO
4 wolframites (e.g., Mg or Mn) and W–O covalent bonds are less compressible than
A–O bonds, the internal stretching modes of WO
6 are the four modes in the high-frequency part of the Raman spectrum (2A
g + 2B
g) plus one A
g mode located near 550 cm
−1 and one A
g mode with a frequency near 400 cm
−1. To facilitate identification by the reader, the six internal modes are identified by an asterisk in
Table 2. Notice that these six modes have pressure coefficients that do not change much from one compound to the other, and are among the largest pressure coefficients (dω/dP). These observations are consistent with the fact that these modes are associated with internal vibrations on the WO
6 octahedron and with the well-known incompressibility of it [
10], explained in the previous section.
The twelve Raman modes not corresponding to internal stretching vibrations of the WO
6 octahedron imply either bending or motions of WO
6 units against the divalent atom
A. These modes are in the low-frequency region and are of particular interest because they are very sensitive to structural symmetry changes. Most of these modes (except to B
g modes) have smaller pressure coefficients than do the internal modes. As expected, all Raman-active modes harden under compression, which indicates that the phase transitions induced by pressure are not associated with soft-mode mechanisms. A similar behavior has been reported for wolframite-type MnWO
4–FeWO
4 solid-solutions [
32].
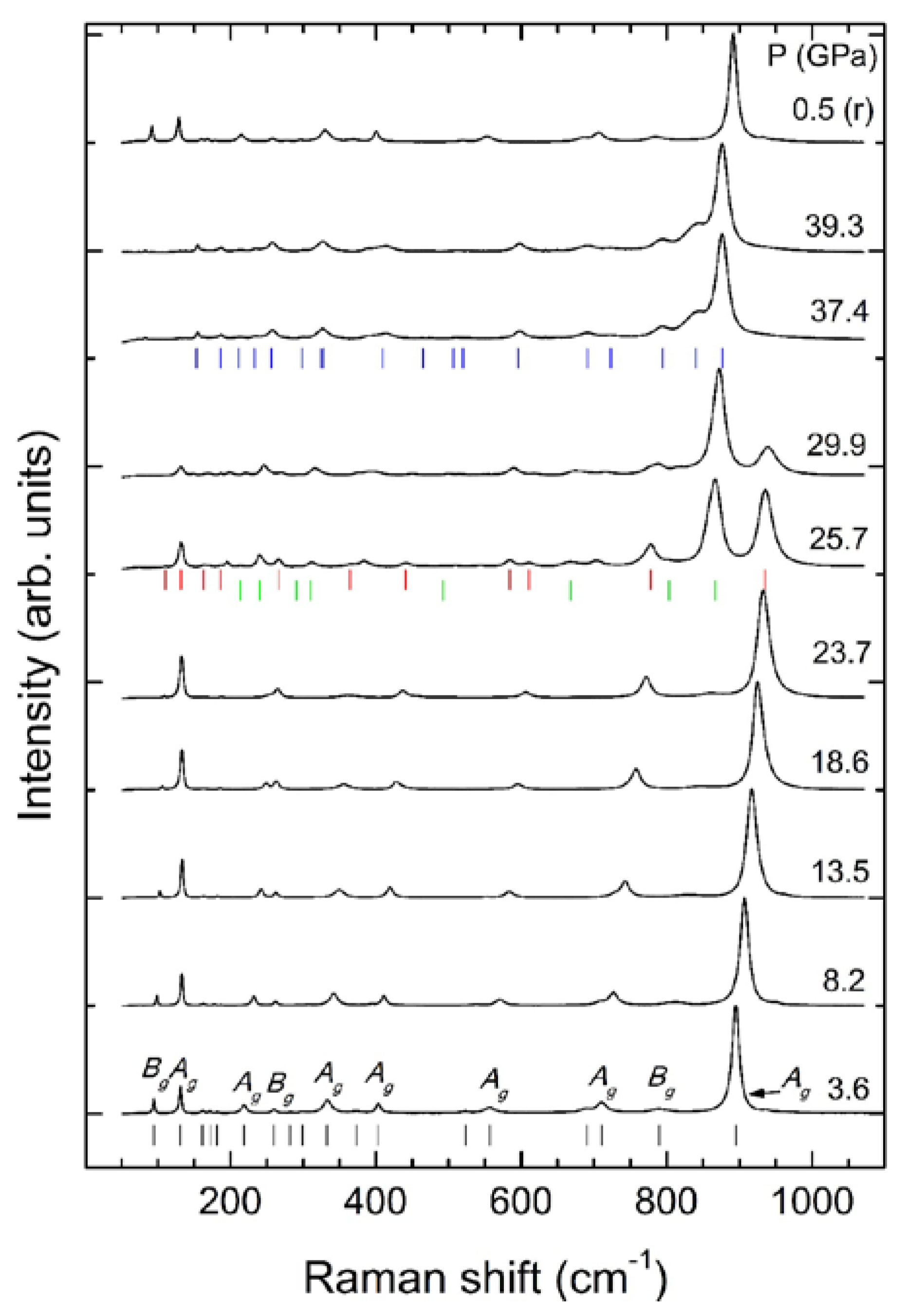
Raman spectroscopy is very sensitive to pressure-driven phase transitions in wolframites. In order to illustrate this, we show in
Figure 6 a selection of Raman spectra measured in MnWO
4 at different pressures. The experiments were carried out in a diamond-anvil cell using neon as a pressure medium to guarantee quasi-hydrostatic conditions [
20]. Clear changes in the Raman spectrum are detected at 25.7 GPa. In particular, new peaks are detected. These changes are indicative of the onset of a structural phase transition. Above 25.7 GPa, gradual changes occur in the Raman spectrum—the Raman modes of the low-pressure wolframite phase only beyond 35 GPa disappear—which indicates the completion of the phase transition. In parallel, the Raman modes of the high-pressure (HP) phase steadily gain intensity from 25.7 to 35 GPa. In
Figure 6, the Raman spectra reported at 37.4 and 39.3 GPa correspond to a single post-wolframite phase. In the same figure, it can be seen that the phase transition is reversible, the Raman spectrum collected at 0.5 GPa under decompression corresponding to the wolframite phase. The number of Raman-active modes detected for this HP phase of MnWO
4 is 18 (the same number as that for the wolframite-type phase). These modes have been assigned in accordance with the crystal structure proposed for the HP phase [
13,
31]. This structure is a triclinic distortion of wolframite, and the same number of Raman-active modes is expected; however, all of them have A
g symmetry in the HP phase.
Table 3 shows the frequencies of the Raman-active modes of the HP phase of MnWO
4 and its pressure coefficients. A detailed discussion of the Raman spectrum of the HP phase can be found elsewhere [
20] and is beyond the scope of this article. Here, we will just mention its most relevant features: (1) The modes are no longer isolated in three groups, which suggests that vibrations cannot be explained with a model that assumes that the WO
6 octahedron is an isolated unit. (2) The most intense mode is the highest frequency mode (
Figure 6), which indicates that it might be associated with a W–O stretching vibration. (3) Interestingly, this mode drops in frequency in comparison to the highest frequency mode in the low-pressure wolframite phase. This is indicative of an increase in W–O bond length evolving towards an effective increasing of the W–O coordination. (4) All the modes of the HP phase harden under compression, but the pressure coefficients of the different modes are smaller than those in the low-pressure phase, which can be explained by the decrease in the compressibility of MnWO
4.
Phase transitions are detected by Raman spectroscopy in other
AWO
4 tungstates from 20 to 30 GPa [
17,
18,
19]. In CdWO
4, ZnWO
4, and MgWO
4 phase coexistence is found between the low- and high-pressure phases in a pressure range of 10 GPa. The Raman spectra of the HP phase of all of the compounds studied up to now resemble very much those of the HP phase of MnWO
4. However, there are small discrepancies that suggest that, even though the HP phase is structurally related to wolframite, there are some differences in their HP phases. These can be clearly seen when comparing the Raman spectra published for the HP phases of CdWO
4 [
18] and MnWO
4 [
20]. In CdWO
4, the phase transition to the post-wolframite structure leads to a doubling of the unit-cell (hence, the formula unit increases from 2 to 4) [
14], with the consequent increase in the number of Raman modes. They have been assigned to 19 A
g + 17 B
g modes. Out of them, only 26 modes have been observed [
14,
18]. The frequency of these modes and the pressure coefficients are summarized in
Table 4. These values agree very well with theoretical predictions for the monoclinic HP phase of CdWO
4 [
18]. The larger number of modes observed in the HP phase of CdWO
4 in comparison with MnWO
4 clearly indicates that the crystal structures of the two compounds are different. In spite of this fact, in the post-wolframite phase of CdWO
4, there is a substantial drop in the frequency of the highest frequency mode (the most intense one), as observed in MnWO
4 [
20]. In the case of CdWO
4, a clear correlation can be established between the frequency drop in this W–O stretching mode and the increase in the tungsten–oxygen coordination number [
13]. Regarding the pressure dependence of the Raman modes in post-wolframite CdWO
4, in
Table 4, it can be seen that, as in MnWO
4, in the HP of CdWO
4 phase the vibrational modes are less affected by compression. In the HP phase of CdWO
4, there are two modes with negative pressure coefficients. The existence of modes with negative pressure slopes might be an indication of structural instability like that observed in related scheelite-type tungstates [
33].
To conclude the discussion on tungstates, we would like to state that, in ZnWO
4 and MgWO
4 [
7,
19], fewer than 18 Raman modes have been found for the HP phase. The frequency distribution of Raman modes more closely resembles that of MnWO
4 than that of CdWO
4. However, no definitive conclusion can be stated on the structure of the HP phase of ZnWO
4 or MgWO
4 only from Raman spectroscopy measurements. Indeed, the accurate assignment of the modes of the HP phases of ZnWO
4 and MgWO
4 requires HP single-crystal experiments similar to those already carried out with MnWO
4 and CdWO
4 [
13,
14].
HP Raman studies have been also carried out for wolframite-type InTaO
4 and InNbO
4 [
15,
16]. In both compounds, the Raman spectrum and its pressure evolution resemble those for the tungstates described above. Again, in the low-pressure phase, the modes that change more under compression are the highest frequency modes, which correspond to internal stretching vibrations of the TaO
6 or NbO
6 octahedron. In these compounds, the phase transition occurs around 15 GPa. At the transition, there is a redistribution of high-frequency modes (which seems to be a fingerprint of a transition to post-wolframite), which involves a drop in frequency in the highest frequency mode and other changes consistent with coordination changes determined by XRD experiments [
15,
16].
5. Electronic Structure and Band Gap
The knowledge of the electronic band structure of wolframite-type compounds is important for the development of the technological applications of these materials. The study of the pressure effects in the band gap has been proven to be an efficient tool for testing the electronic band structure of materials. The first efforts to accurately determine the band structure of wolframites were made by Abraham et al. [
34]. These authors focused on CdWO
4, comparing it with the better known scheelite-type CdMoO
4. By means of density-functional theory calculations, they found that the lowermost conduction bands of CdWO
4 are controlled by the crystal-field-splitting of the 5
d bands of tungsten (slightly hybridized with O 2
p states). On the other hand, the upper part of the valence band is mainly constituted by O 2
p states (slightly hybridized with W 5
d states). As a consequence, ZnWO
4, CdWO
4, and MgWO
4 have very similar band-gap energy
Eg. Accurate values of
Eg have been determined by means of optical-absorption measurements,
Eg being equal to 3.98, 4.02, and 4.06 eV in ZnWO
4, CdWO
4, and MgWO
4, respectively [
22]. On the contrary, MnWO
4 is known to have a considerably smaller band gap,
Eg = 2.37 eV [
22]. This distinctive behavior is the consequence of the contribution of the Mn 3
d5 orbitals to the states near the Fermi energy. Basically, Mg (3
s2), Zn (3
d10), or Cd (4
d10) have filled electronic shells and therefore do not contribute either to the top of the valence band or the bottom of the conduction band. However, in MnWO
4, Mn (3
d5) contributes to the top of the valence band and the bottom of the conduction band, reducing the band-gap energy in comparison to ZnWO
4, CdWO
4, and MgWO
4. A similar behavior to that of MnWO
4 is expected for NiWO
4, CoWO
4, and FeWO
4 due to the presence of Ni 3
d8, Co 3
d7, and Fe 3
d6 states. Therefore, among the wide dispersion of values reported for NiWO
4 (2.28 eV <
Eg < 4.5 eV) [
35], those in the lowest limit appear to be the most realistic values. Notice that the above conclusions are in agreement with the fact that CuWO
4, a distorted wolframite, in which Cu 3
d9 states contribute to the top of the conduction band, has a band-gap energy of 2.3 eV [
36]. Another relevant difference between the band structure of the first and second group of compounds is that it necessarily implies that ZnWO
4, CdWO
4, and MgWO
4 are direct band gap materials, but the other wolframites are indirect band-gap materials [
22].
An analogous behavior to that of the wolframite-tungstates is expected for isomorphic molybdates. In this case, the states near the Fermi level will be basically dominated by MoO
42− [
37], so wolframite-type molybdates should have slightly smaller
Eg than the tungsten-containing counterpart [
38]. Unfortunately, less efforts have been dedicated to molybdates than to tungstates. However, the above stated hypothesis has been confirmed in the case of wolframite-type ZnMoO
4, which has
Eg = 3.22 eV [
39]. A similar value is expected for MgMoO
4, ruling out estimations that range from 4.5 to 5.5 eV [
40]. On the other hand, the band-gap energy of 2.2 eV determined for NiMoO
4 [
41] is fully consistent with the conclusions extracted from MnWO
4. In this case, the Ni 3
d8 states will be responsible of the reduction of its
Eg in comparison with ZnMoO
4.
InTaO
4 and InNbO
4 are promising materials for photocatalytic water splitting applications [
42]. Both materials have been found to be indirect band-gap semiconductors, with
Eg = 3.79 eV in InTaO
4 [
16]. In this compound, the O 2
p states, with a small amount of mixing of the In 4
d states, dominate the upper part of the valence band, and Ta 5
d states and In 5
s dominate the lower conduction bands. An analogous situation is expected for InNbO
4, with the only difference being that Nb 4
d contributes to the bottom of the conduction band and the Ta 5
d states do not. This fact makes
Eg slightly smaller in InNbO
4 (nearly 3.4 eV) than in InTaO
4 [
43]. An explanation for this phenomenon comes from the larger Pauling’s electronegativity of Nb (1.6) in comparison with Ta (1.5) [
44]. Within a basic tight-binding approach,
Eg is proportional to the overlap integral between the wave functions of atoms. Since Ta has a smaller Pauling’s electronegativity than Nb, in a tantalate, the electron transfer from Ta to neighboring oxygen atoms is expected to be larger than the electron transfer from Nb to the neighboring oxygen atoms in a niobate. Therefore, the superposition of wave functions of Ta and O is larger than that of Nb and O, resulting in a larger
Eg in InTaO
4 than in InNbO
4. The same argument can be used to justify the fact that, systematically, molybdates have a smaller
Eg than tungstates, which has been stated in the previous paragraph.
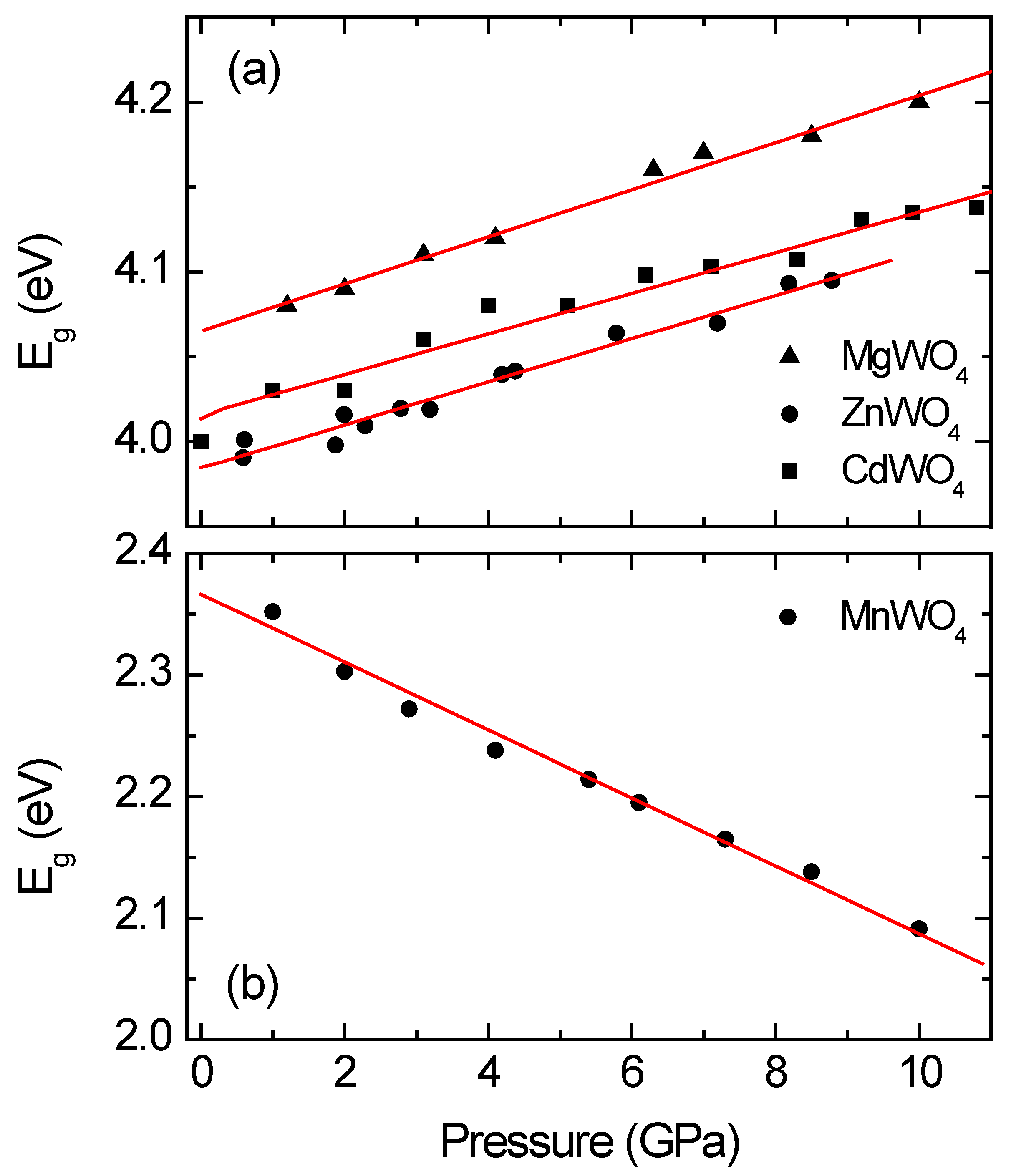
We shall discuss now the influence of pressure on the band structure of wolframites. We will first focus on wolframite-type tungstates. The pressure dependence of
Eg has been determined from optical-absorption experiments up to 10 GPa for CdWO
4, MgWO
4, MnWO
4, and ZnWO
4 [
22]. The experiments were carried out on single-crystal samples, under quasi-hydrostatic conditions up to a maximum pressure which is far away from the transition pressure. The obtained results are summarized in
Figure 7. There it can be seen that MnWO
4 has a very distinctive behavior. For MgWO
4, ZnWO
4, and CdWO
4, the pressure dependence of
Eg can be represented by a linear function with a positive slope close to d
Eg/d
P = 13 meV/GPa [
22]. However, a different pressure dependence is followed by MnWO
4, in which
Eg redshifts at −22 meV/GPa [
22]. An explanation to the different behavior of
Eg in MnWO
4 comes from the contribution of Mn 3
d5 states to the bottom of the conduction band. Whereas for MgWO
4, ZnWO
4, and CdWO
4, under compression the bottom of the conduction band goes up in energy, the contribution of Mn states makes it to go down [
22]. On the other hand, for the four compounds, the top of the valence band keeps the same energy. This is translated into an increase in
Eg for MgWO
4, ZnWO
4, and CdWO
4 and a decrease in
Eg for MnWO
4. The same behavior in MnWO
4 has been reported for triclinic wolframite-related CuWO
4 [
35] and can be predicted for CoWO
4, FeWO
4, and NiWO
4.
The same arguments used to explain the HP behavior of the band-gap in wolframites are useful for understanding the related scheelite-type and monazite-type oxides [
45]. An example of it are scheelite-type CaMoO
4 (CaWO
4) and PbMoO
4 (PbWO
4) [
46,
47]. Another example of it is monazite-type SrCrO
4 and PbCrO
4 [
48,
49]. In all these compounds, the Pb-containing compounds have a smaller
Eg than their isomorphic compounds. This is due to the contribution of Pb 6
s (6
p) states to the top (bottom) of the valence (conduction) band. In addition, the Pb states make the band gap to close under compression. As a consequence, d
Eg/d
P is negative in the Pb containing compounds, but has the opposite sign in the other compounds.
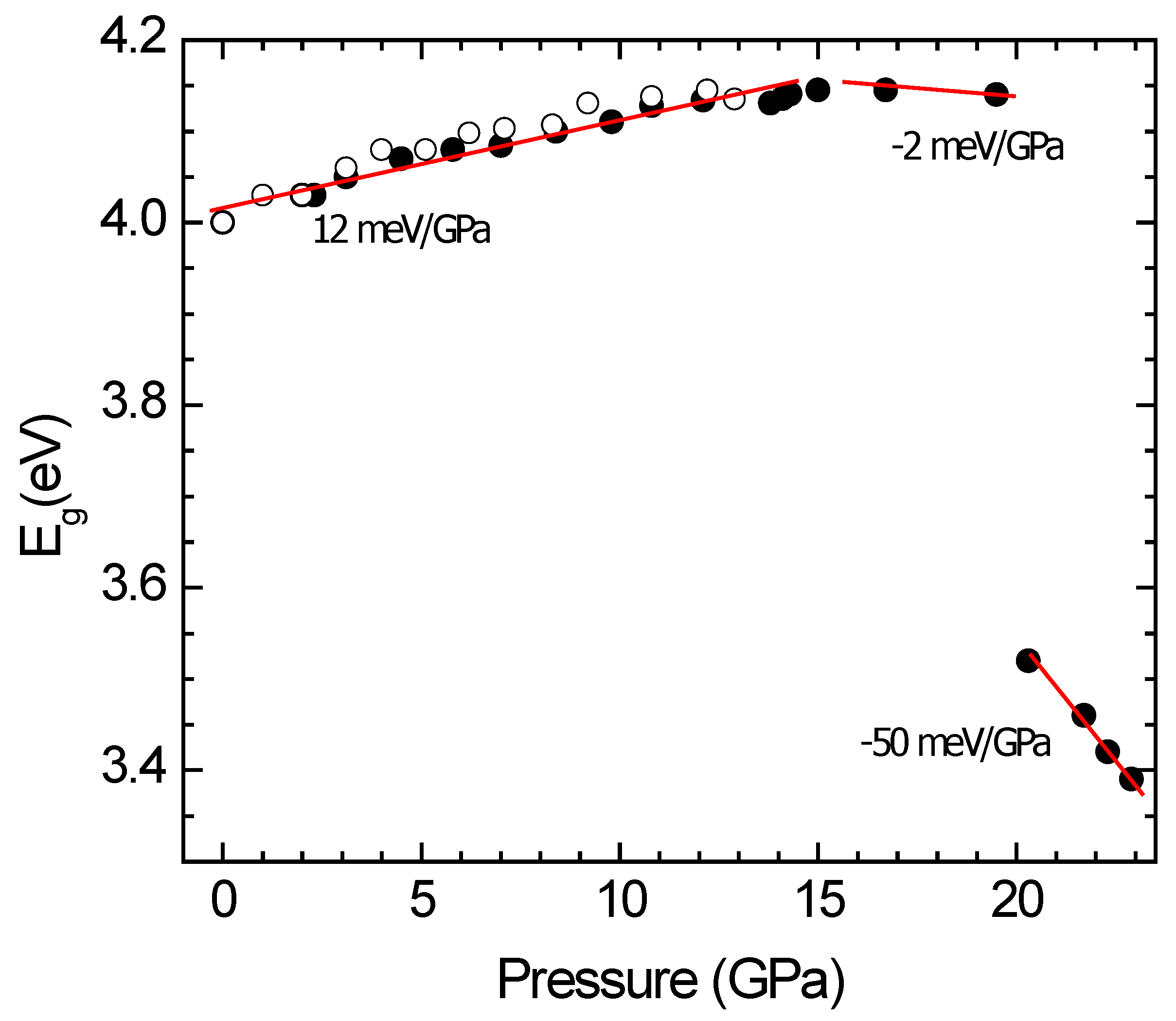
Studies beyond the pressure range of stability have been carried out for CdWO
4 [
14]. In this compound optical-absorption studies under quasi-hydrostatic conditions have been performed up to 23 GPa. The pressure dependence determined of
Eg is shown in
Figure 8. As described above, in the first compression steps,
Eg increases with pressure. However, at 16.9 GPa, the behavior of
Eg changes, moving to lower energies under compression up to 19.5 GPa. This change is caused by a band crossing of the direct and indirect band gaps [
14], this result being consistent with Raman and XRD measurements. Beyond 19.5 GPa, a drastic color change occurs in the sample as the result of a sharp band gap reduction to ∼3.5 eV [
14]. The changes are triggered by the onset of the structural phase transition described in a previous section. In the HP phase of CdWO
4 Eg decreases with pressure. Regarding the slope change observed at 16.9 GPa, it is a consequence of a direct to indirect band-gap transition caused by the modification of the electronic band structure under compression [
14,
50]. Such band crossing has also been observed in other wolframites, such as InTaO
4 [
15]. This phenomenon should influence not only the band gap but also other band-structure parameters, such as the effective masses, having a strong influence in transport properties [
51], an issue that deserves to be explored in the future.
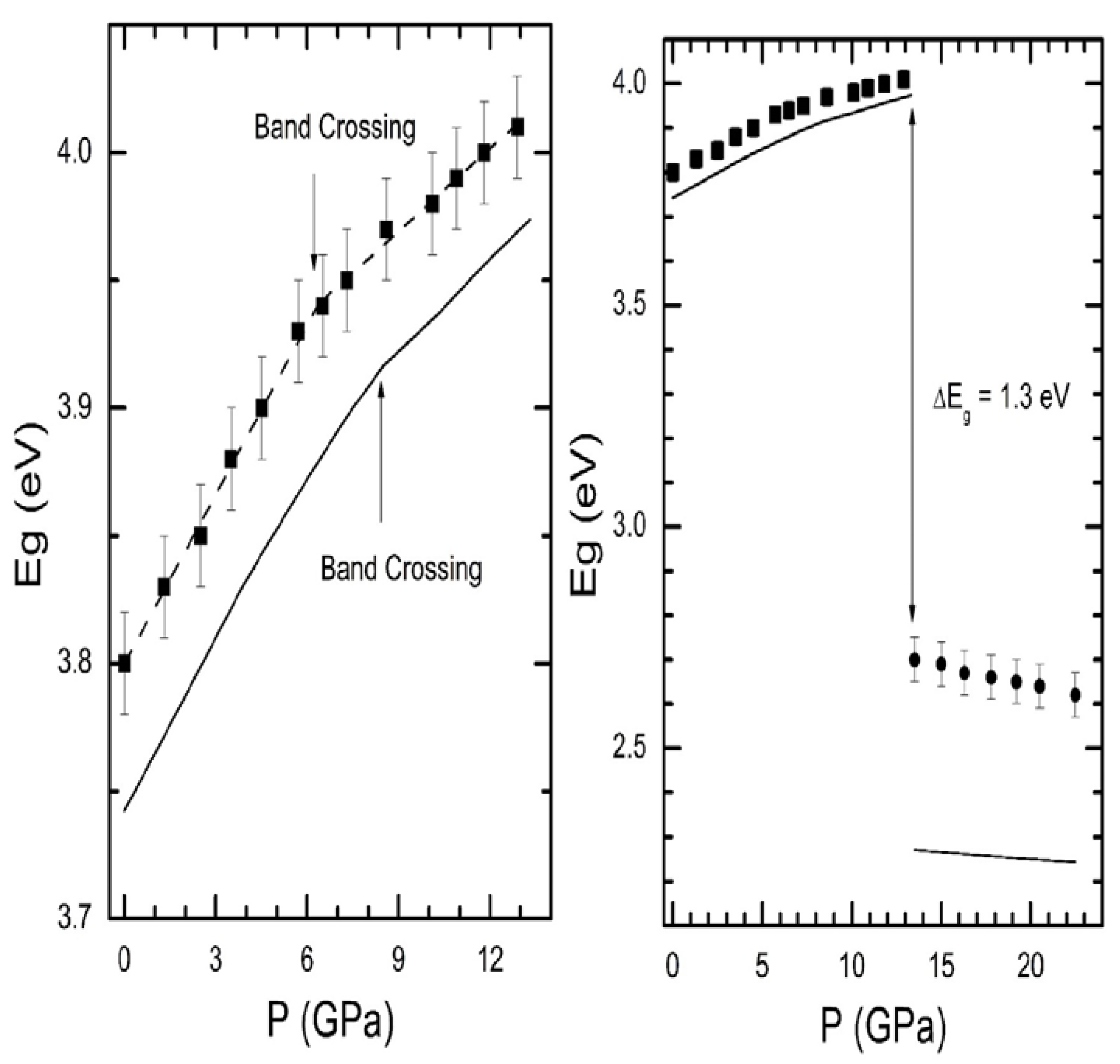
Let us discuss now the case of InTaO
4. In this compound,
Eg has been experimentally determined up to 23 GPa [
15]. Calculations have been also carried out [
15]. The results are shown in
Figure 9. Calculations underestimate the value of
Eg, which is typical of density-functional theory; however, they nicely reproduce the pressure dependence of
Eg. In the figure, it can be seen that, when pressure is increased, there is a blueshift of
Eg with a change of the d
Eg/d
P around 5 GPa. As in CdWO
4, this singularity occurs due to a band crossing [
15], which in the case of InTaO
4 is triggered by changes induced in the top of the valence band by pressure. In particular, at around 5 GPa, the maximum of the valence band changes from the Y point of the Brillouin zone to the Z point. On the other hand, when increasing the pressure beyond 13 GPa, InTaO
4 changes from colorless to yellow [
15]. The change in color is correlated to a band-gap collapse and is associated with a structural phase transition found by Raman and XRD experiments [
15]. The HP phase has been found to have a direct band gap (the low-pressure wolframite has an indirect gap). In contrast with the low-pressure phase, in the HP phase
Eg redshifts with pressure. This is a consequence of the fact that, under compression, the valence band shifts slightly faster towards high energies than the conduction band.
To close this section, we would like to comment on the possible pressure-induced metallization of wolframites. It has been suggested, based upon resistivity measurements, that ternary oxides related to wolframite might metallize through band overlapping at relative low pressures (12–30 GPa) [
52,
53]. So far, no evidence of metallization has been detected for all the studied wolframites, either in the pressure range of stability of the low-pressure phase or in the post-wolframite phases up to the maximum pressure achieved in experiments (45 GPa in ZnWO
4 and CdWO
4) [
17,
18]. In particular, the sample darkening associated with a semiconductor–metal transition has never been detected in any wolframite under compression. In addition, density-functional theory calculations also exclude the possibility of metallization in the wolframite and post-wolframite phases [
14,
15]. One of the reasons preventing metallization is the robustness of the WO
6 (MO
6, NbO
6, and TaO
6) octahedron, which is a less compressible polyhedral unit within the crystal structure. As a consequence, the application of high pressures is not enough to increase the overlap between the electronic wave-functions of transition metals (W, Mo, Nb, and Ta) and oxygen atoms to broaden the electronic bands and create the eventual delocalization of the electrons requested for metallization [
54]. The fact that in distorted wolframite CuWO
4 a HP phase transition takes place without any significant reduction of the Jahn–Teller distortion [
55] suggests that this compound is the best candidate for metallization driven by band overlap under compression.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}