Conformational Flexibility of Proteins Involved in Ribosome Biogenesis: Investigations via Small Angle X-ray Scattering (SAXS)

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Circular Dichroism Spectroscopy (CD)
2.3. Macromolecular Crystallography (MX)
2.3.1. Crystallization
2.3.2. Data Collection, Data Processing, Model Building, and Refinement
2.4. Small Angle X-ray Scattering (SAXS)
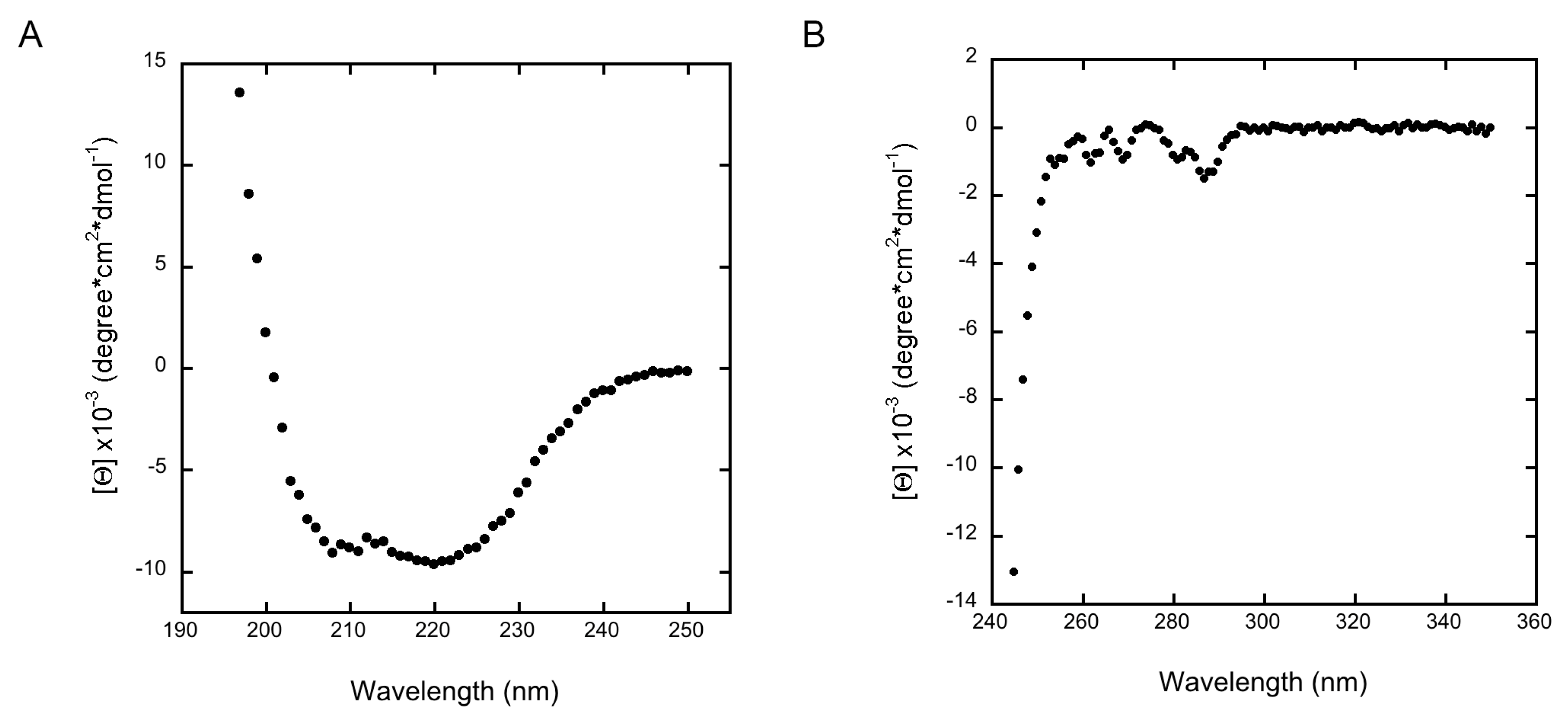
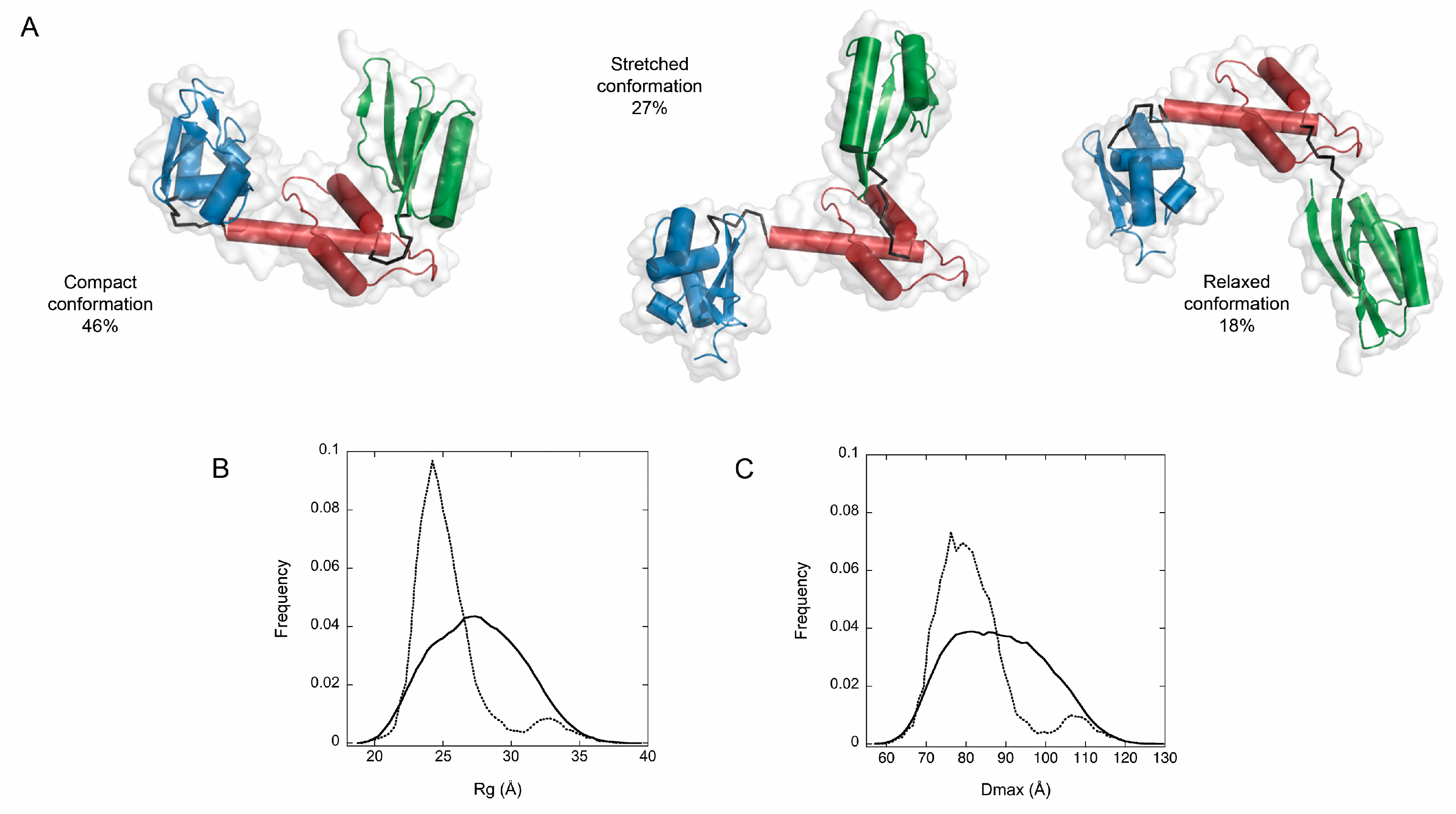
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rambo, R.P.; Tainer, J.A. Super-resolution in solution X-ray scattering and its applications to structural systems biology. Annu. Rev. Biophys. 2013, 42, 415–441. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, C.E.; Spilotros, A.; Schwemmer, F.; Graewert, M.A.; Kikhney, A.; Jeffries, C.M.; Franke, D.; Mark, D.; Zengerle, R.; Cipriani, F.; et al. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Crystallogr. 2015, 48, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef] [PubMed]
- Bernado, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef] [PubMed]
- Boura, E.; Rozycki, B.; Herrick, D.Z.; Chung, H.S.; Vecer, J.; Eaton, W.A.; Cafiso, D.S.; Hummer, G.; Hurley, J.H. Solution structure of the ESCRT-I complex by small-angle X-ray scattering, EPR, and FRET spectroscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 9437–9442. [Google Scholar] [CrossRef] [PubMed]
- Boura, E.; Rozycki, B.; Chung, H.S.; Herrick, D.Z.; Canagarajah, B.; Cafiso, D.S.; Eaton, W.A.; Hummer, G.; Hurley, J.H. Solution structure of the ESCRT-I and -II supercomplex: Implications for membrane budding and scission. Structure 2012, 20, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Chalupska, D.; Eisenreichova, A.; Rozycki, B.; Rezabkova, L.; Humpolickova, J.; Klima, M.; Boura, E. Structural analysis of phosphatidylinositol 4-kinase IIIbeta (PI4KB)—14-3-3 protein complex reveals internal flexibility and explains 14-3-3 mediated protection from degradation in vitro. J. Struct. Biol. 2017, 200, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Panse, V.G.; Johnson, A.W. Maturation of eukaryotic ribosomes: Acquisition of functionality. Trends. Biochem. Sci. 2010, 35, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Tschochner, H.; Hurt, E. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends. Cell Biol. 2003, 13, 255–263. [Google Scholar] [CrossRef]
- Venema, J.; Tollervey, D. Ribosome synthesis in Saccharomyces cerevisiae. Annu. Rev. Genet. 1999, 33, 261–311. [Google Scholar] [CrossRef] [PubMed]
- Woolford, J.L., Jr.; Baserga, S.J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 2013, 195, 643–681. [Google Scholar] [CrossRef] [PubMed]
- Ting, Y.H.; Lu, T.J.; Johnson, A.W.; Shie, J.T.; Chen, B.R.; Kumar, S.S.; Lo, K.Y. Bcp1 Is the Nuclear Chaperone of Rpl23 in Saccharomyces cerevisiae. J. Biol. Chem. 2017, 292, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Wyler, E.; Wandrey, F.; Badertscher, L.; Montellese, C.; Alper, D.; Kutay, U. The beta-isoform of the BRCA2 and CDKN1A(p21)-interacting protein (BCCIP) stabilizes nuclear RPL23/uL14. FEBS Lett. 2014, 588, 3685–3691. [Google Scholar] [CrossRef] [PubMed]
- Gartmann, M.; Blau, M.; Armache, J.P.; Mielke, T.; Topf, M.; Beckmann, R. Mechanism of eIF6-mediated inhibition of ribosomal subunit joining. J. Biol. Chem. 2010, 285, 14848–14851. [Google Scholar] [CrossRef] [PubMed]
- Menne, T.F.; Goyenechea, B.; Sanchez-Puig, N.; Wong, C.C.; Tonkin, L.M.; Ancliff, P.J.; Brost, R.L.; Costanzo, M.; Boone, C.; Warren, A.J. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 2007, 39, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Marquez, A.; Gijsbers, A.; de la Mora, E.; Sanchez-Puig, N. Defective Guanine Nucleotide Exchange in the Elongation Factor-like 1 (EFL1) GTPase by Mutations in the Shwachman-Diamond Syndrome Protein. J. Biol. Chem. 2015, 290, 17669–17678. [Google Scholar] [CrossRef] [PubMed]
- Gijsbers, A.; Garcia-Marquez, A.; Luviano, A.; Sanchez-Puig, N. Guanine nucleotide exchange in the ribosomal GTPase EFL1 is modulated by the protein mutated in the Shwachman-Diamond Syndrome. Biochem. Biophys. Res. Commun. 2013, 437, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Weis, F.; Giudice, E.; Churcher, M.; Jin, L.; Hilcenko, C.; Wong, C.C.; Traynor, D.; Kay, R.R.; Warren, A.J. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat. Struct. Mol. Biol. 2015, 22, 914–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savchenko, A.; Krogan, N.; Cort, J.R.; Evdokimova, E.; Lew, J.M.; Yee, A.A.; Sanchez-Pulido, L.; Andrade, M.A.; Bochkarev, A.; Watson, J.D.; et al. The Shwachman-Bodian-Diamond syndrome protein family is involved in RNA metabolism. J. Biol. Chem. 2005, 280, 19213–19220. [Google Scholar] [CrossRef] [PubMed]
- Shammas, C.; Menne, T.F.; Hilcenko, C.; Michell, S.R.; Goyenechea, B.; Boocock, G.R.; Durie, P.R.; Rommens, J.M.; Warren, A.J. Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman-Diamond Syndrome. J. Biol. Chem. 2005, 280, 19221–19229. [Google Scholar] [CrossRef] [PubMed]
- Gorrec, F. The MORPHEUS protein crystallization screen. J. Appl. Crystallogr. 2009, 42, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Allan, D.R.; Collins, S.P.; Evans, G.; Hall, D.; McAuley, K.; Owen, R.L.; Sorensen, T.; Tang, C.C.; von Delft, F.; Wagner, A.; et al. Status of the crystallography beamlines at Diamond Light Source. Eur. Phys. J. Plus 2015, 130, 50. [Google Scholar] [CrossRef]
- Mueller, M.; Wang, M.; Schulze-Briese, C. Optimal fine phi-slicing for single-photon-counting pixel detectors. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Krojer, T.; Pike, A.C.; von Delft, F. Squeezing the most from every crystal: The fine details of data collection. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall III, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Liu, J.; Shen, Z. Inhibition of G1 to S cell cycle progression by BCCIP beta. Cell Cycle 2004, 3, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Guo, X.; Meng, X.; Liu, J.; Allen, C.; Wray, J.; Nickoloff, J.A.; Shen, Z. The BRCA2-interacting protein BCCIP functions in RAD51 and BRCA2 focus formation and homologous recombinational repair. Mol. Cell. Biol. 2005, 25, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Slabinski, L.; Jaroszewski, L.; Rychlewski, L.; Wilson, I.A.; Lesley, S.A.; Godzik, A. XtalPred: A web server for prediction of protein crystallizability. Bioinformatics 2007, 23, 3403–3405. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.L.; Waterman, D.G.; Koonin, E.V.; Walters, A.D.; Chong, J.P.; Isupov, M.N.; Lebedev, A.A.; Bunka, D.H.; Stockley, P.G.; Ortiz-Lombardia, M.; et al. Conformational flexibility and molecular interactions of an archaeal homologue of the Shwachman-Bodian-Diamond syndrome protein. BMC Struct. Biol. 2009, 9, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinier, A. La diffraction des rayons X aux très petits angles: Application à l’étude de phénomènes ultramicroscopiques. Ann. Phys. 1939, 11, 161–237. [Google Scholar] [CrossRef]
- Glatter, O. A new method for the evaluation of small-angle scattering data. J. Appl. Cryst. 1977, 10, 415–421. [Google Scholar] [CrossRef]
- Svergun, D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999, 76, 2879–2886. [Google Scholar] [CrossRef]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Tuukkanen, A.T.; Kleywegt, G.J.; Svergun, D.I. Resolution of ab initio shapes determined from small-angle scattering. IUCrJ 2016, 3, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef]
- Tria, G.; Mertens, H.D.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I.; Barberato, C.; Koch, M.H.J. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Finch, A.J.; Hilcenko, C.; Basse, N.; Drynan, L.F.; Goyenechea, B.; Menne, T.F.; Gonzalez Fernandez, A.; Simpson, P.; D’Santos, C.S.; Arends, M.J.; et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011, 25, 917–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | |
|---|---|
| Space group | P1 |
| Cell dimensions | |
| a, b, c (Å) | 33.81, 44.52, 54.71 |
| α, β, γ (°) | 75.07, 84.52, 69.72 |
| Resolution (Å) | 36.71–1.73 (1.77–1.73) |
| Rsym | 0.178 (0.648) |
| I/σI | 1.7 (1.1) |
| Completeness (%) | 94.6 (90.9) |
| Redundancy | 1.8 (1.7) |
| Refinement | |
| Resolution (Å) | 1.9 |
| No. reflections | 21715 |
| Rwork/Rfree | 0.197/0.232 |
| No. atoms | |
| Protein Ligand (PEG) | 1861 7 |
| Water | 81 |
| B-factors | |
| Protein Ligand (PEG) | 46.05 61.63 |
| Water | 47.25 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 0.824 |
| Data Collection Parameters | ||
|---|---|---|
| BeamLine | B21, Diamond Light Source, Harwell (UK) | |
| Detector | Pilatus 2M | |
| Beam size | 0.2 × 0.2 mm | |
| Energy | 12.4 keV | |
| Sample-to-detector distance (mm) | 4014 | |
| q range (A−1) | 0.0038–0.42 | |
| Exposure time (s) | 10 and 3 (in HPLC mode) | |
| Temperature (K) | 293 | |
| Structural Parameters | ||
| Bcp1 | AfSBDS | |
| Concentration range (mg/mL) | 1.1–3.70 | 5.0 in HPLC mode |
| q Interval for Fourier inversion (Å−1) | 0.014–0.314 | 0.016–0.306 |
| Rg [from P(r)] (Å) | 27.03 ± 0.51 | 25.51 ± 0.02 |
| Rg [from Guiner approximation] (Å) | 29.02 ± 1.49 | 24.73 ± 2.12 |
| sRg limits [from Guiner approximation] | 0.58–1.49 | 0.27–1.29 |
| Dmax (Å) | 102.00 | 87.00 |
| Porod volume estimate (nm3) | 68.83 | 32.84 |
| DAMMIF excluded volume (nm3) | 75.04 | 46.76 |
| Molecular Mass (kDa) | ||
| From Porod (× 0.53) | 36.50 | 17.41 |
| From excluded volume (× 0.5) | 37.52 | 23.00 |
| From sequence | 27.72 | 26.56 |
| Modelling | ||
| Ambiguity | 0.0 (unique) | 2.9 (highly ambiguous) |
| Resolution (Å) | 29 ± 2 | 30 ± 2 |
| Software Employed | ||
| Primary data reduction | DAWN pipeline (Diamond Light Source, UK) | |
| Data processing | ScÅtter v3.1q | |
| Ab initio modelling | DAMMIF/DAMAVER | |
| Validation and averaging | DAMAVER/DAMCLUST | |
| Computation of model intensities | CRYSOL | |
| Flexibility | EOM | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siliqi, D.; Foadi, J.; Mazzorana, M.; Altamura, D.; Méndez-Godoy, A.; Sánchez-Puig, N. Conformational Flexibility of Proteins Involved in Ribosome Biogenesis: Investigations via Small Angle X-ray Scattering (SAXS). Crystals 2018, 8, 109. https://doi.org/10.3390/cryst8030109
Siliqi D, Foadi J, Mazzorana M, Altamura D, Méndez-Godoy A, Sánchez-Puig N. Conformational Flexibility of Proteins Involved in Ribosome Biogenesis: Investigations via Small Angle X-ray Scattering (SAXS). Crystals. 2018; 8(3):109. https://doi.org/10.3390/cryst8030109
Chicago/Turabian StyleSiliqi, Dritan, James Foadi, Marco Mazzorana, Davide Altamura, Alfonso Méndez-Godoy, and Nuria Sánchez-Puig. 2018. "Conformational Flexibility of Proteins Involved in Ribosome Biogenesis: Investigations via Small Angle X-ray Scattering (SAXS)" Crystals 8, no. 3: 109. https://doi.org/10.3390/cryst8030109
APA StyleSiliqi, D., Foadi, J., Mazzorana, M., Altamura, D., Méndez-Godoy, A., & Sánchez-Puig, N. (2018). Conformational Flexibility of Proteins Involved in Ribosome Biogenesis: Investigations via Small Angle X-ray Scattering (SAXS). Crystals, 8(3), 109. https://doi.org/10.3390/cryst8030109