Effects of Empirical Dispersion Energy on the Geometrical Parameters and Relative Energy of a Salicylideneaniline Molecular Switch in the Solid State




Abstract
:1. Introduction
2. Computational Aspects
3. Results and Discussion
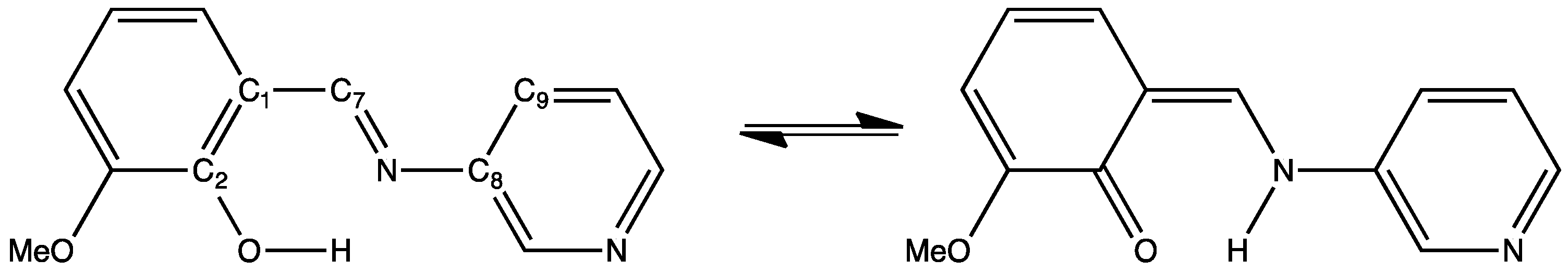
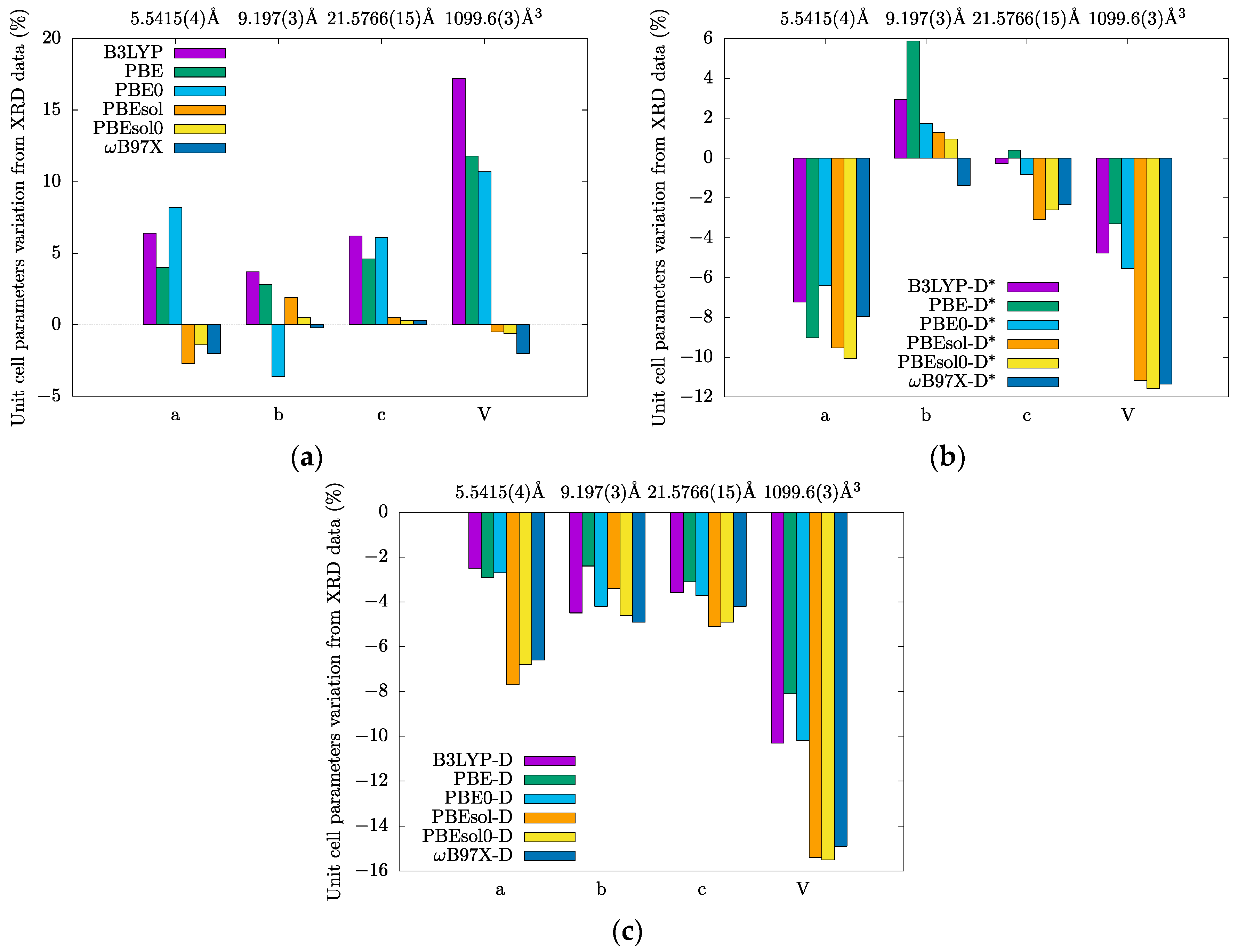
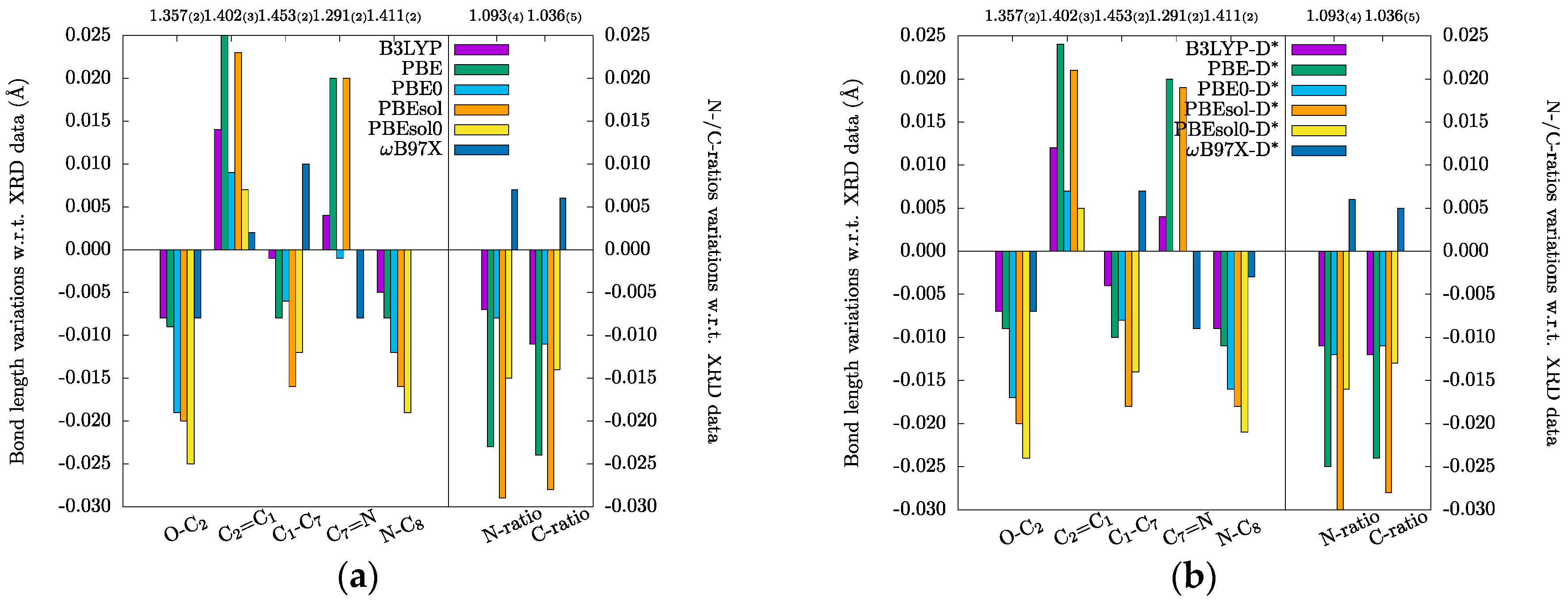
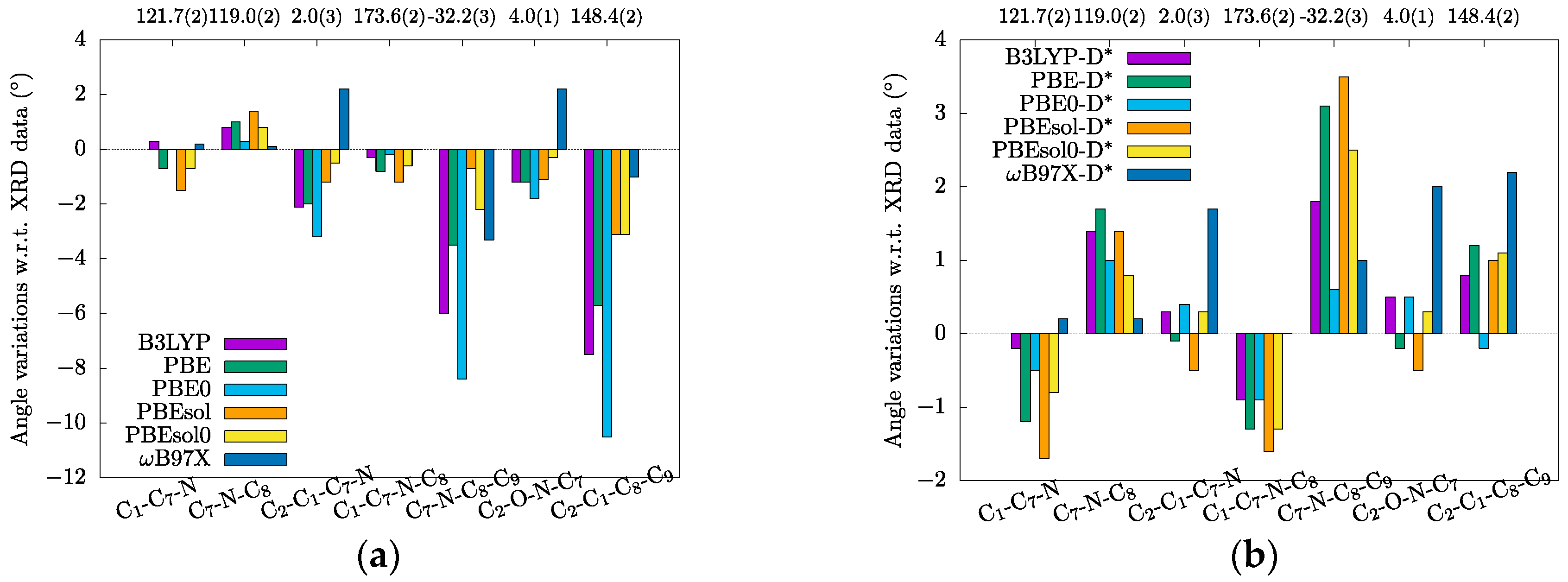
3.1. Crystal Structures and Molecular Geometries
3.2. Keto–Enol Energies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Crano, J.C.; Guglielmetti, R.J. Organic Photochromic and Thermochromic Compounds; Springer: New York, NY, USA, 1998. [Google Scholar]
- Kawata, S.; Kawata, Y. Three-dimensional optical data storage using photochromic materials. Chem. Rev. 2000, 100, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Hadjoudis, E.; Mavridis, I.M. Photochromism and thermochromism of Schiff bases in the solid state: Structural aspects. Chem. Soc. Rev. 2004, 33, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Amimoto, K.; Kawato, T. Photochromism of organic compounds in the crystal state. J. Photochem. Photobiol. C Photochem. Rev. 2005, 6, 207–226. [Google Scholar] [CrossRef]
- Feringa, B.L.; Browne, W.R. Molecular Switches, 2nd ed.; WILEY-VCH Press: Weinheim, Germany, 2011. [Google Scholar]
- Antonov, L. Tautomerism: Methods and Theories; Wiley-VCH Press: Weinheim, Germany, 2013. [Google Scholar]
- Antonov, L. Tautomerism: Concepts and Applications in Science and Technology; WILEY-VCH Press: Weinheim, Germany, 2016. [Google Scholar]
- Padalkar, V.S.; Seki, S. Excited-state intramolecular proton-transfer (ESIPT)-inspired solid state emitters. Chem. Soc. Rev. 2016, 45, 169–202. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, M.T.; Gooch, J.; Zubieta, J.; Korter, T.M. Evaluation of range-corrected density functionals for the simulation of pyridinium-containing molecular crystals. J. Phys. Chem. A 2016, 120, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Quertinmont, J.; Carletta, A.; Tumanov, N.A.; Leyssens, T.; Wouters, J.; Champagne, B. Assessing density functional theory approaches for predicting the structure and relative energy of salicylideneaniline molecular switches in the solid state. J. Phys. Chem. C 2017, 121, 6898–6908. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Schimka, L.; Harl, J.; Kresse, G. Improved hybrid functional for solids: The HSEsol functional. J. Chem. Phys. 2011, 134, 024116. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Civalleri, B.; Zicovich-Wilson, C.M.; Valenzano, L.; Ugliengo, P. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 2008, 10, 405–410. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Erba, A.; Zicovich-Wilson, C.M.; Civalleri, B.; Casassa, S.; Maschio, L.; Ferrabone, M.; De La Pierre, M.; D’Arco, P.; et al. CRYSTAL14: A program for the ab initio investigation of crystalline solids. Int. J. Quantum Chem. 2014, 114, 1287–1317. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward chemical accuracy in the computation of NMR shieldings: The PBE0 model. Chem. Phys. Lett. 1998, 298, 113–119. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [PubMed]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Carletta, A.; Buol, X.; Leyssens, T.; Champagne, B.; Wouters, J. Polymorphic and isomorphic cocrystals of a n-salicylidene-3-aminopyridine with dicarboxylic acids: tuning of solid-state photo- and thermochromism. J. Phys. Chem. C 2016, 120, 10001–10008. [Google Scholar] [CrossRef]
- Carletta, A.; Spinelli, F.; D’Agostino, S.; Ventura, B.; Chierotti, M.R.; Gobetto, R.; Wouters, J.; Grepioni, F. Halogen-bond effects on the thermo- and photochromic behaviour of anil-based molecular co-crystals. Chem.-A Eur. J. 2017, 23, 5317–5329. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | DFT | DFT-D2 | DFT-D* |
|---|---|---|---|
| B3LYP | 189.0 (17.2%) | −112.9 (−10.3%) | −52.3 (−4.8%) |
| PBE | 129.4 (11.8%) | −89.3 (−8.1%) | −36.2 (−3.3%) |
| PBE0 | 117.3 (10.7%) | −112.3 (−10.5%) | −61.1 (−5.6%) |
| PBEsol | −5.2 (−0.5%) | −169.3 (−15.4%) | −122.9 (−11.2%) |
| PBEsol0 | −7.0 (−0.6%) | −170.5 (−15.5%) | −127.2 (−11.6%) |
| ωB97X 1 | −21.9 (−2.0%) | −163.4 (−14.9%) | −124.9 (−11.4%) |
| Method | DFT | DFT-D2 | DFT-D* |
|---|---|---|---|
| B3LYP | 8.3 (3%) | 2.7 (25%) | 3.4 (20%) |
| PBE | 2.6 (26%) | −2.5 (73%) | −1.3 (63%) |
| PBE0 | 11.0 (1%) | 4.5 (14%) | 5.3 (11%) |
| PBEsol | 1.0 (40%) | −2.7 (75%) | −1.9 (68%) |
| PBEsol0 | 7.1 (5%) | 3.4 (20%) | 4.0 (17%) |
| ωB97X 1 | 12.9 (1%) | 10.2 (2%) | 9.9 (2%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quertinmont, J.; Leyssens, T.; Wouters, J.; Champagne, B. Effects of Empirical Dispersion Energy on the Geometrical Parameters and Relative Energy of a Salicylideneaniline Molecular Switch in the Solid State. Crystals 2018, 8, 125. https://doi.org/10.3390/cryst8030125
Quertinmont J, Leyssens T, Wouters J, Champagne B. Effects of Empirical Dispersion Energy on the Geometrical Parameters and Relative Energy of a Salicylideneaniline Molecular Switch in the Solid State. Crystals. 2018; 8(3):125. https://doi.org/10.3390/cryst8030125
Chicago/Turabian StyleQuertinmont, Jean, Tom Leyssens, Johan Wouters, and Benoît Champagne. 2018. "Effects of Empirical Dispersion Energy on the Geometrical Parameters and Relative Energy of a Salicylideneaniline Molecular Switch in the Solid State" Crystals 8, no. 3: 125. https://doi.org/10.3390/cryst8030125
APA StyleQuertinmont, J., Leyssens, T., Wouters, J., & Champagne, B. (2018). Effects of Empirical Dispersion Energy on the Geometrical Parameters and Relative Energy of a Salicylideneaniline Molecular Switch in the Solid State. Crystals, 8(3), 125. https://doi.org/10.3390/cryst8030125