Insight into Physical and Thermodynamic Properties of X3Ir (X = Ti, V, Cr, Nb and Mo) Compounds Influenced by Refractory Elements: A First-Principles Calculation

Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Structural Properties
3.2. Elastic Constants
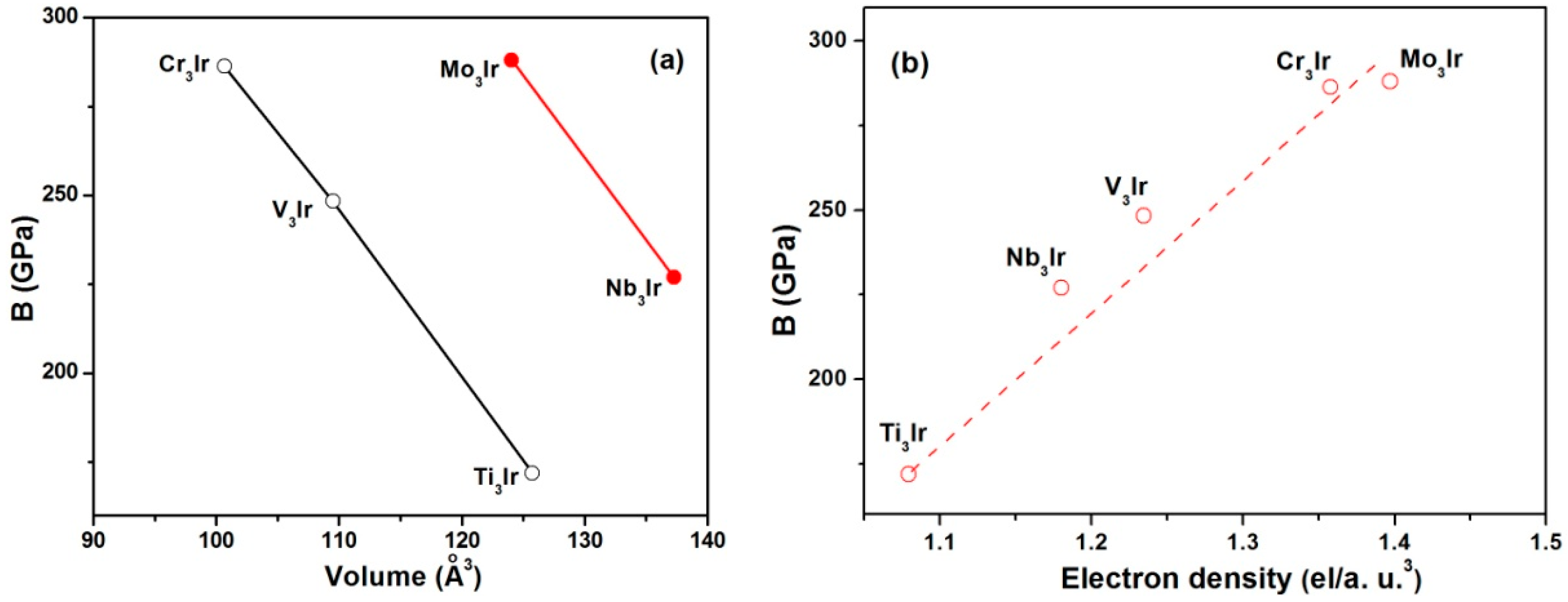
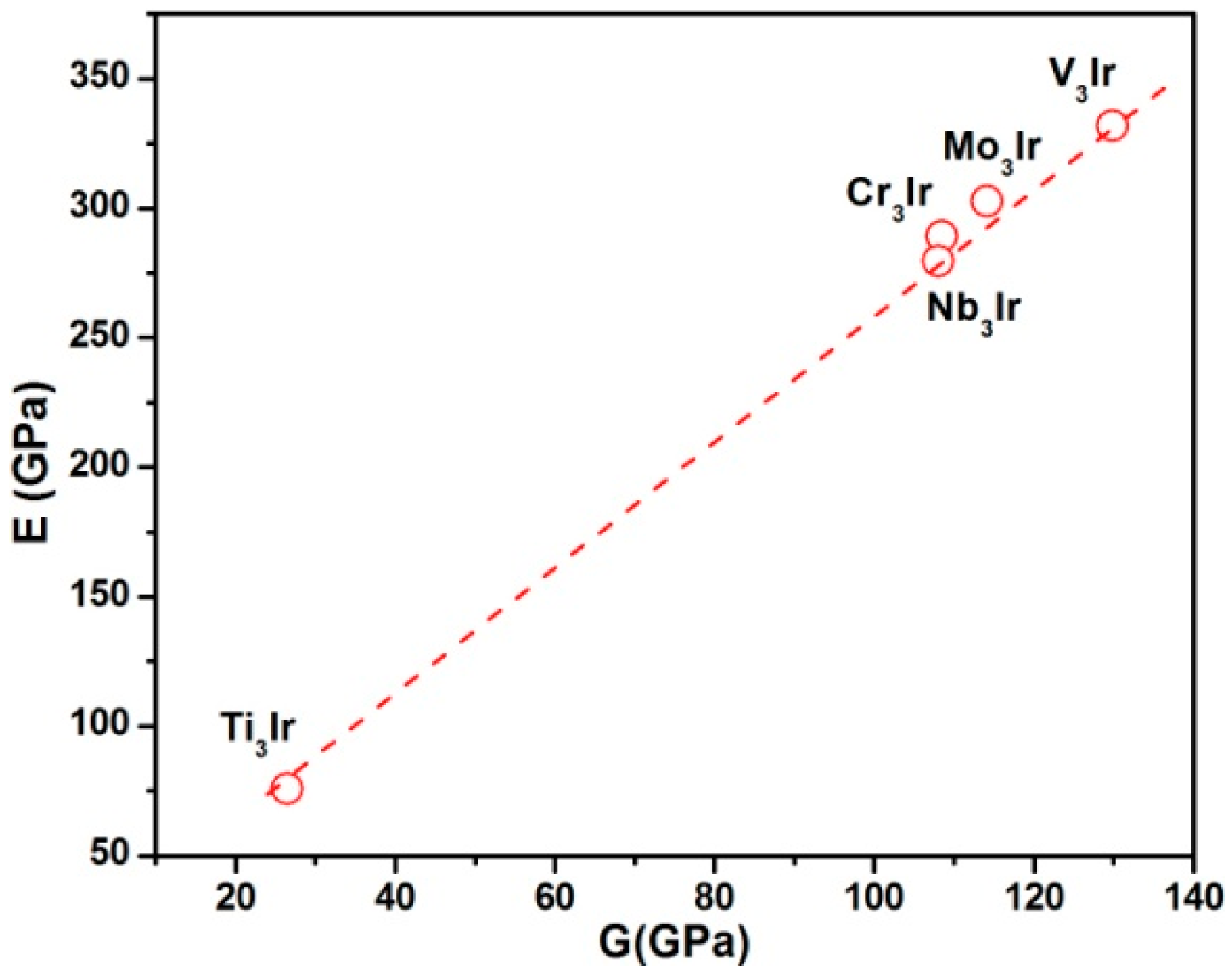
3.3. Elastic Properties
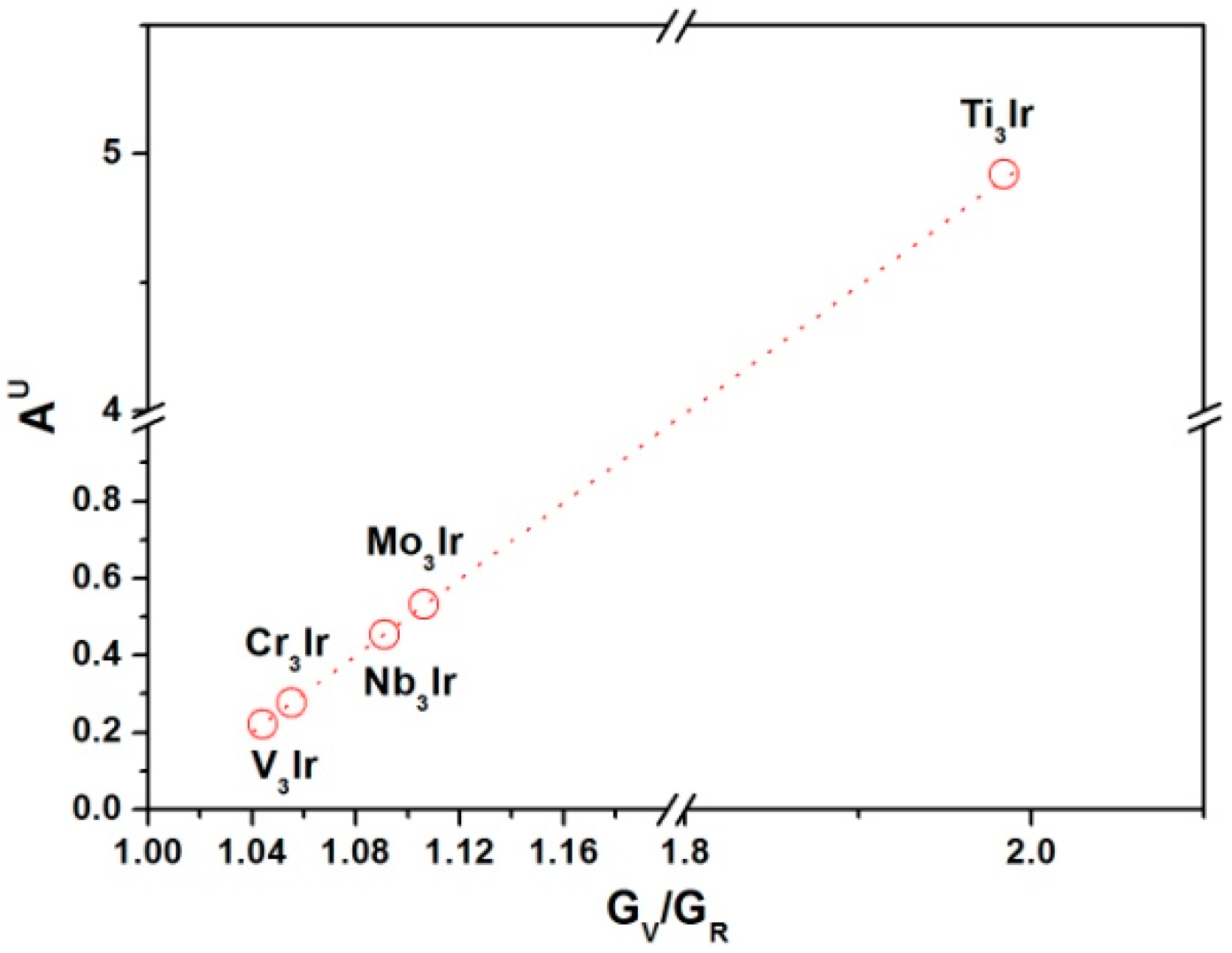
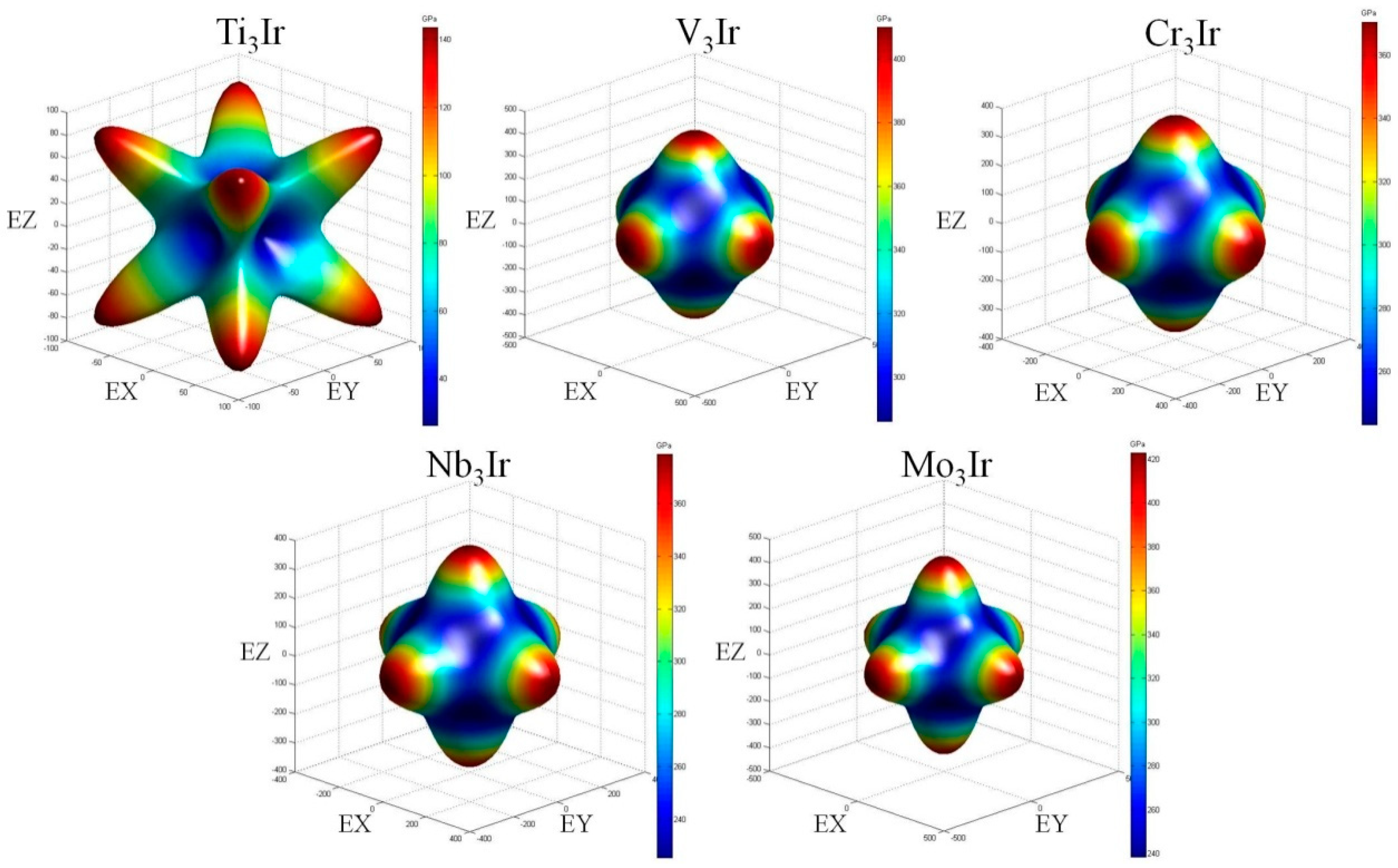
3.4. Elastic Anisotropy
3.5. Anisotropic Sound Velocity and Debye Temperature
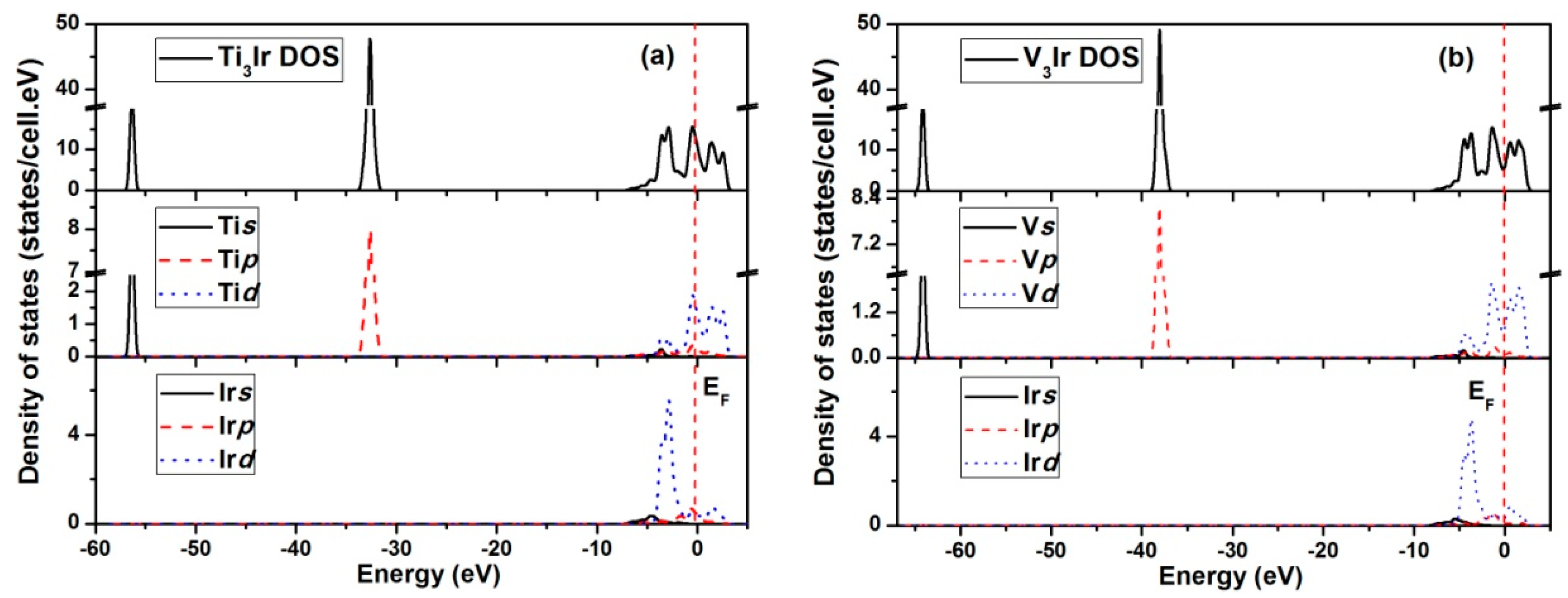
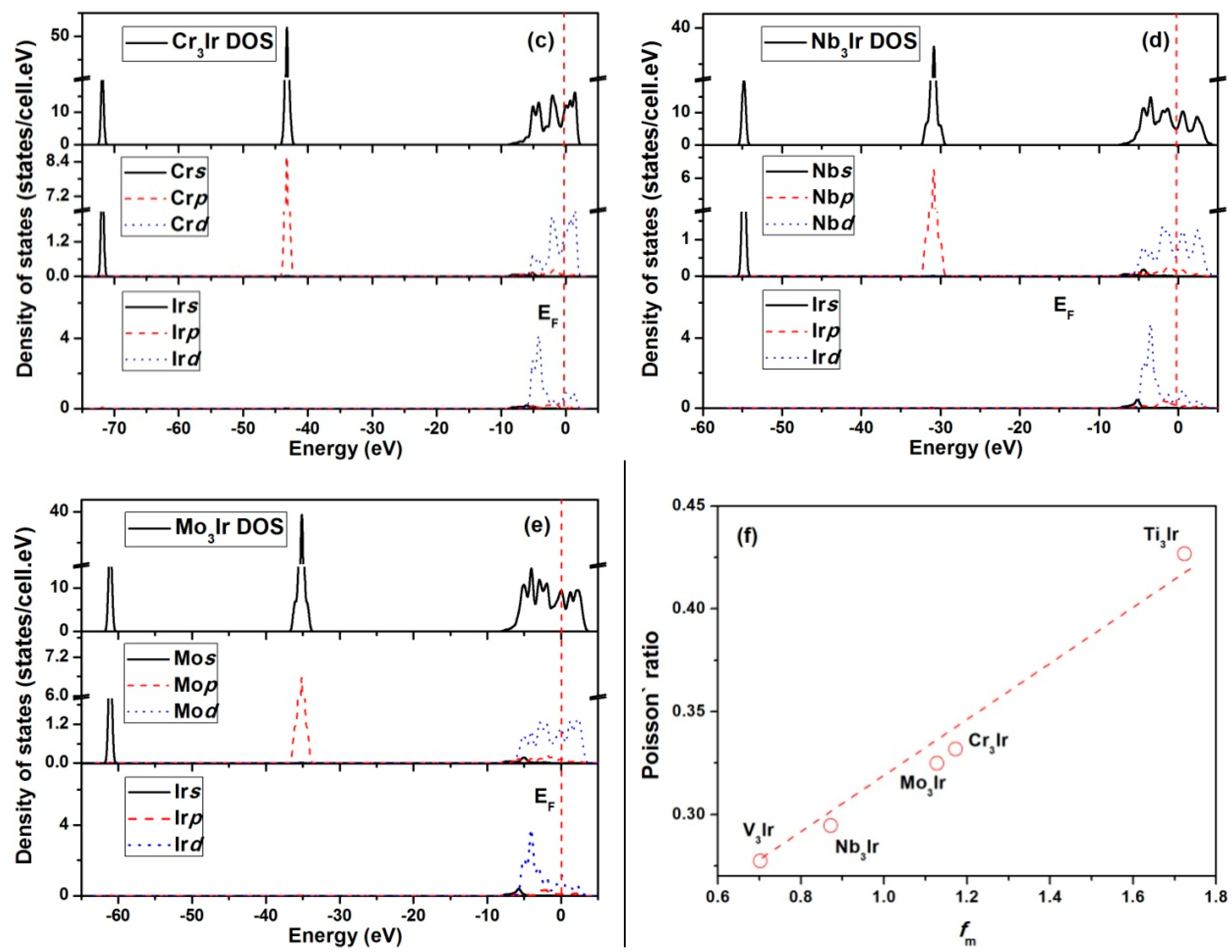
3.6. Electronic Structures
- kB represents the Boltzmann constant (k = 1.381 × 10−23 J/K);
- T represents the absolute temperature;
- Df represents the DOS value at the Fermi level;
- nm and ne represent the thermally excited electrons and valence electron density of the cell, respectively;
- ne is calculated by ne = N/Vcell (N represents the total number of valence electrons; Vcell represents the cell volume).
4. Conclusions
- (1)
- Using the GGA method to structurally optimized the unit cell, smaller calculation deviations for lattice constants were achieved as compared to those achieved using the LDA method.
- (2)
- The calculated bulk moduli exhibited the increasing sequence of Ti3Ir < Nb3Ir < V3Ir < Cr3Ir < Mo3Ir. Furthermore, the bulk moduli showed a linear relationship with electron densities. The Young’s modulus showed a linear dependence on shear modulus following the order of Ti3Ir < Nb3Ir < Cr3Ir < Mo3Ir < V3Ir.
- (3)
- Based on the discussions on the Cauchy pressure, Poisson’s ratio and B/G ratio, the ductile essence was found to be enhanced in the order of V3Ir < Nb3Ir < Mo3Ir < Cr3Ir < Ti3Ir.
- (4)
- For X3Ir compounds, the extent of the elastic anisotropy for X3Ir obeyed the increasing sequence of V3Ir < Cr3Ir < Nb3Ir < Mo3Ir < Ti3Ir via the analyses of the universal anisotropic indexes and 3D surface constructions.
- (5)
- The Debye temperatures obtained for Ti3Ir, V3Ir, Cr3Ir and Nb3Ir were all in good agreement with the results from experiments. Such good compliance proved the superior quality of our calculations of the structural and elastic properties, since the computation of Debye temperature is concerned with both structural and elastic parameters.
- (6)
- The calculated electronic structures for X3Ir compounds showed similar features in the DOS spectra. Furthermore, the metallicity of the compounds was calculated, and was correlated with the Poisson’s ratios. This indicated that a compound with higher metallicity in its bonds should possess better ductility.
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Yamaguchi, M.; Inui, H.; Ito, K. High-temperature structural intermetallics. Acta Mater. 2000, 48, 307–322. [Google Scholar] [CrossRef]
- Yu, X.; Yamabe-Mitarai, Y.; Ro, Y.; Harada, H. New developed quaternary refractory superalloys. Intemetallics 2000, 8, 619–622. [Google Scholar] [CrossRef]
- Yamabe-Mitarai, Y.; Gu, Y.; Huang, C.; Völkl, R.; Harada, H. Platinum-group-metal-based intermetallics as high-temperature structural materials. JOM 2004, 56, 34–39. [Google Scholar] [CrossRef]
- Yamabe-Mitari, Y.; Ro, Y.; Maruko, T.; Harada, H. Microstructure dependence of strength of Ir-base refractory superalloys. Intermetallics 1999, 7, 49–58. [Google Scholar] [CrossRef]
- Yamabe-Mitarai, Y.; Ro, Y.; Nakazawa, S. Temperature dependence of the flow stress of Ir-based L12 intermetallics. Intermetallics 2001, 9, 423–429. [Google Scholar] [CrossRef]
- Terada, Y.; Ohkubo, K.; Miura, S.; Sanchez, J.M.; Mohri, T. Thermal conductivity and thermal expansion of Ir3X (X = Ti, Zr, Hf, V, Nb, Ta) compounds for high-temperature applications. Mater. Chem. Phys. 2003, 80, 385–390. [Google Scholar] [CrossRef]
- Chen, K.; Zhao, L.R.; Tse, J.S. Ab initio study of elastic properties of Ir and Ir3X compounds. J. Appl. Phys. 2003, 93, 2414–2417. [Google Scholar] [CrossRef]
- Liu, N.; Wang, X.; Wan, Y. Firstprinciple calculations of elastic and thermodynamic properties of Ir3Nb and Ir3V with L12 structure under high pressure. Intermetallics 2015, 66, 103–110. [Google Scholar] [CrossRef]
- Pan, Y.; Lin, Y.; Xue, Q.; Ren, C.; Wang, H. Relationship between Si concentration and mechanical properties of Nb-Si compounds: A first-principles study. Mater. Des. 2016, 89, 676–683. [Google Scholar] [CrossRef]
- Papadimitriou, I.; Utton, C.; Scott, A.; Tsakiropoulos, P. Ab Initio Study of Binary and Ternary Nb3(X,Y) A15 Intermetallic Phases (X,Y = Al, Ge, Si, Sn). Metall. Mater. Trans. A 2015, 46, 566–576. [Google Scholar] [CrossRef]
- Papadimitriou, I.; Utton, C.; Tsakiropoulos, P. Ab initio investigation of the intermetallics in the Nb-Sn binary system. Acta Mater. 2015, 86, 23–33. [Google Scholar] [CrossRef]
- Papadimitriou, I.; Utton, C.; Tsakiropoulos, P. Ab initio investigation of the Nb-Al system. Comput. Mater. Sci. 2015, 107, 116–121. [Google Scholar] [CrossRef]
- Chihi, T.; Fatmi, M. Theoretical prediction of the structural, elastic, electronic and thermodynamic properties of V3M (M = Si, Ge and Sn) compounds. Superlatt. Microstruc. 2012, 52, 697–703. [Google Scholar] [CrossRef]
- Jarlborg, T.; Junod, A.; Peter, M. Electronic structure, superconductivity, and spin fluctuations in the A15 compounds A3B: A =V, Nb; B=Ir, Pt, Au. Phys. Rev. B 1983, 27, 1558–1567. [Google Scholar] [CrossRef]
- Paduani, C.; Kuhnen, C.A. Band structure calculations in isoelectronic V3B compounds: B=Ni, Pd and Pt. Solid State Commun. 2010, 150, 1303–1307. [Google Scholar] [CrossRef]
- Staudenmann, J.L.; DeFacio, B.; Testardi, L.R.; Werner, S.A.; Flükiger, R.; Muller, J. Debye classes in A15 compounds. Phys. Rev. B 1981, 24, 6446. [Google Scholar] [CrossRef]
- Meschel, S.V.; Kleppa, O.J. The standard enthalpies of formation of some intermetallic compounds of transition metals by high temperature direct synthesis calorimetry. J. Alloy. Compd. 2006, 415, 143–149. [Google Scholar] [CrossRef]
- Paduani, C.; Kuhnen, C.A. Electronic structure of A15-type compounds: V3Co, V3Rh, V3Ir and V3Os. Eur. Phys. J. B 2009, 69, 331–336. [Google Scholar] [CrossRef]
- Paduani, C. Electronic properties of the A-15 Nb-based intermetallics Nb3(Os,Ir,Pt,Au). Solid State Commun. 2007, 144, 352–356. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Zeitschrift fuer Kristallographie 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef]
- Shanno, D.F. Conditioning of quasi-Newton methods for function minimization. Math. Comput. 1970, 24, 647–656. [Google Scholar] [CrossRef]
- Fischer, T.H.; Almlof, J. General Methods for Geometry and Wave Function Optimization. J. Chem. Phys. 1992, 96, 9768–9774. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Ti3Ir, ICSD No. 641115.
- V3Ir, ICSD No. 104591.
- Cr3Ir, ICSD No. 102780.
- Nb3Ir, ICSD No. 640833.
- Mo3Ir, ICSD No.600761.
- Rajagopalan, M.; Gandhi, R.R. First principles study of structural, electronic, mechanical and thermal properties of A15 intermetallic compounds Ti3X (X = Au, Pt, Ir). Phys. B 2012, 407, 4731–4734. [Google Scholar] [CrossRef]
- Paduani, C. Structural and electronic properties of the A-15 compounds Nb3Rh and Nb3Ir. Phys. B 2007, 393, 105–109. [Google Scholar] [CrossRef]
- Subhashree, G.; Sankar, S.; Krithiga, R. Superconducting properties of Mo3Os,Mo3Pt,Mo3Ir from first principle calculations. Mod. Phys. Lett. B 2014, 28, 1450233. [Google Scholar] [CrossRef]
- Afaq, A.; Rizwan, M.; Bakar, A. Computational investigations of XMgGa (X = Li, Na) half Heusler compounds for thermo-elastic and vibrational properties. Phys. B 2019, 554, 102–106. [Google Scholar] [CrossRef]
- Heciri, D.; Belkhir, H.; Belghit, R.; Bouhafs, B.; Khenata, R.; Ahmed, R.; Bouhemadou, A.; Ouahrani, T.; Wang, X.; Omrani, S.B. Insight into the structural, elastic and electronic properties of tetragonal inter-alkali metal chalcogenides CsNaX (X = S, Se, and Te) from first-principles calculations. Mater. Chem. Phys. 2019, 221, 125–137. [Google Scholar] [CrossRef]
- He, D.G.; Lin, Y.C.; Jiang, X.Y.; Yin, L.X.; Wang, L.H.; Wu, Q. Dissolution mechanisms and kinetics of δ phase in an aged Ni-based superalloy in hot deformation process. Mater. Des. 2018, 156, 262–271. [Google Scholar] [CrossRef]
- Pettifor, D.G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 1992, 8, 345–349. [Google Scholar] [CrossRef]
- Fatima, B.; Chouhan, S.S.; Acharya, N.; Sanyal, S.P. Theoretical prediction of the electronic structure, bonding behavior and elastic moduli of scandium intermetallics. Intermetallics 2014, 53, 129–139. [Google Scholar] [CrossRef]
- Sundareswari, M.; Ramasubramanian, S.; Rajagopalan, M. Elastic and thermodynamical properties of A15 Nb3X (X = Al,Ga,In,Sn and Sb) compounds-First principles DFT study. Solid State Commun. 2010, 150, 2057–2060. [Google Scholar] [CrossRef]
- Han, Y.; Wu, Y.; Li, T.; Khenata, R.; Yang, T.; Wang, X. Electronic, Magnetic, Half-Metallic, and Mechanical Properties of a New Equiatomic Quaternary Heusler Compound YRhTiGe: A First-Principles Study. Materials 2018, 11, 797. [Google Scholar] [CrossRef]
- Chen, D.; Chen, Z.; Wu, Y.; Wang, M.; Ma, N.; Wang, H. First-principles investigation of mechanical, electronic and optical properties of Al3Sc intermetallic compound under pressure. Comput. Mater. Sci. 2014, 91, 165–172. [Google Scholar] [CrossRef]
- Salma, M.U.; Rahman, M.A. Study of structural, elastic, electronic, mechanical, optical and thermodynamic properties of NdPb3 intermetallic compound: DFT based calculations. Comput. Condens. Matter 2018, 15, 42–47. [Google Scholar] [CrossRef]
- Luan, X.; Qin, H.; OrcID, F.L.; Dai, Z.; Yi, Y.; Li, Q. The Mechanical Properties and Elastic Anisotropies of Cubic Ni3Al from First Principles Calculations. Crystals 2018, 8, 307. [Google Scholar] [CrossRef]
- Sultana, F.; Uddin, M.M.; Ali, M.A.; Hossain, M.M.; Naqib, S.H.; Islam, A.K.M.A. First principles study of M2InC (M = Zr, Hf and Ta) MAX phases: The effect of M atomic species. Results Phys. 2018, 11, 869–876. [Google Scholar] [CrossRef]
- Chen, S.; Sun, Y.; Duan, Y.H.; Huang, B.; Peng, M.J. Phase stability, structural and elastic properties of C15-type Laves transition-metal compounds MCo2 from first-principles calculations. J. Alloy. Compd. 2015, 630, 202–208. [Google Scholar] [CrossRef]
- Li, C.X.; Duan, Y.H.; Hu, W.-C. Electronic structure, elastic anisotropy, thermal conductivity and optical properties of calcium apatite Ca5(PO4)3X (X = F, Cl or Br). J. Alloy. Compd. 2015, 619, 66–77. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, C.H.; Li, R.Z.; Shen, J.; Chen, N.X. Site preference and alloying effect on elastic properties of ternary B2 RuAl-based alloys. Intermetallics 2014, 51, 24–29. [Google Scholar] [CrossRef]
- Jacob, K.T.; Raj, S.; Rannesh, L. Vegard’s law: A fundamental relation or an approximation? Inter. J. Mater. Res. 2007, 98, 776–779. [Google Scholar] [CrossRef]
- Li, C.; Wu, P. Correlation of Bulk Modulus and the Constituent Element Properties of Binary Intermetallic Compounds. Chem. Mater. 2001, 13, 4642–4648. [Google Scholar] [CrossRef]
- Chen, D.; Chen, Z.; Wu, Y.; Wang, M.; Ma, N.; Wang, H. First-principles study of mechanical and electronic properties of TiB compound under pressure. Intermetallics 2014, 52, 64–71. [Google Scholar] [CrossRef]
- Zhong, S.Y.; Chen, Z.; Wang, M.; Chen, D. Structural, elastic and thermodynamic properties of Mo3Si and Mo3Ge. Eur. Phys. J. B 2016, 89, 6. [Google Scholar] [CrossRef]
- Lebga, N.; Daoud, S.; Sun, X.W.; Bioud, N.; Latreche, A. Mechanical and Thermophysical Properties of Cubic Rock-Salt AlN Under High Pressure. J. Electr. Mater. 2018, 47, 3430–3439. [Google Scholar] [CrossRef]
- Fu, H.; Li, D.; Peng, F.; Gao, T.; Cheng, X. Ab initio calculations of elastic constants and thermodynamic properties of NiAl under high pressures. Comput. Mater. Sci. 2008, 44, 774–778. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Niu, H.Y.; Chen, X.Q.; Liu, P.T.; Xing, W.W.; Cheng, X.Y.; Li, D.Z.; Li, Y.Y. Extra-electron induced covalent strengthening and generalization of intrinsic ductile-to-brittle criterion. Sci. Rep. 2012, 2, 718. [Google Scholar] [CrossRef]
- Han, C.; Chai, C.; Fan, Q.; Yang, J.; Yang, Y. Structural, Electronic, and Thermodynamic Properties of Tetragonal t-SixGe3-xN4. Materials 2018, 11, 397. [Google Scholar] [CrossRef]
- Belghit, R.; Belkhir, H.; Kadri, M.T.; Heciri, D.; Bououdin, M.; Ahuja, R. Structural, elastic, electronic and optical properties of novel antiferroelectric KNaX (X = S, Se, and Te) compounds: First principles study. Phys. B 2018, 545, 18–29. [Google Scholar] [CrossRef]
- Duan, Y.H.; Sun, Y.; Peng, M.J.; Zhou, S.G. Anisotropic elastic properties of the Ca-Pb compounds. J. Alloy. Compd. 2014, 595, 14–21. [Google Scholar] [CrossRef]
- Vajeeston, P.; Ravindran, P.; Fjellvag, H. Prediction of structural, lattice dynamical, and mechanical properties of CaB2. RSC Adv. 2012, 2, 11687–11694. [Google Scholar] [CrossRef]
- Haque, E.; Hossain, M.A. First-principles study of elastic, electronic, thermodynamic, and thermoelectric transport properties of TaCoSn. Results Phys. 2018, 10, 458–465. [Google Scholar] [CrossRef]
- Junod, A.; Flukiger, R.; Muller, J. Supraconductivite et chaleur specifique dans les alliages A15 a base de titane. J. Phys. Chem. Solids 1976, 37, 27–31. [Google Scholar] [CrossRef]
- Spitzli, P. Chaleur spécifique d’alliages de structure A 15. Physik der kondensierten Materie 1971, 13, 22–58. [Google Scholar]
- Junod, A.; Bischel, D.; Muller, J. Eliashberg inversion of superconducting state thermodynamics. Helv. Phys. Acta 1979, 52, 580. [Google Scholar] [CrossRef]
- Flükiger, R.; Heiniger, F.; Junod, A.; Muller, J.; Spitzli, P.; Staudenmann, J.L. Chaleur specifique et supraconductivite dans des alliages de structure A 15 a base de chrome. J. Phys. Chem. Solids 1971, 32, 459–463. [Google Scholar] [CrossRef]
- Morin, F.J.; Maita, J.P. Specific Heats of Transition Metal Superconductors. Phys. Rev. 1963, 129, 1115. [Google Scholar] [CrossRef]
- Misawa, S. The 3-Dimensional Fermi Liquid Description for the Iron-Based Superconductors. J. Low Temp. Phys. 2018, 190, 45–66. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Xiao, B.; Min, T.; Fan, Z.; Ma, S.; Xu, L. Theoretical study on the stability, elasticity, hardness and electronic structures of W-C binary compounds. J. Alloy. Compd. 2010, 502, 28–37. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | a0 (Å) | aexp (Å) | Calculated Deviation (%) | Density (g/cm3) |
|---|---|---|---|---|
| Ti3Ir | 5.010 a | 5.012 c | −0.041 a | 8.872 a |
| 4.901 b | −2.223 b | 9.479 b | ||
| V3Ir | 4.7842 a | 4.7876 d | −0.072 a | 10.463 a |
| 4.6913 b | −2.012 b | 11.099 b | ||
| Cr3Ir | 4.652 a | 4.685 e | −0.712 a | 11.489 a |
| 4.651 b | −0.732 b | 11.496 b | ||
| Nb3Ir | 5.1585 a | 5.135 f | 0.457 a | 11.394 a |
| 5.0777 b | −1.116 b | 11.946 b | ||
| Mo3Ir | 4.9874 a | 4.9703 g | 0.344 a | 12.986 a |
| 4.9199 b | −1.014 b | 13.387 b |
| Compounds | Cij | C12–C44 (GPa) | B (GPa) | G (GPa) | E (GPa) | v | B/G | ||
|---|---|---|---|---|---|---|---|---|---|
| C11 (GPa) | C44 (GPa) | C12 (GPa) | |||||||
| Ti3Ir | 183.8 | 52.8 | 166.0 | 114.2 | 171.9 | 26.5 | 75.7 | 0.427 | 6.483 |
| 207.2 a | 48.8 a | 153.1 a | 104.3 a | 171.1 a | 38.5 a | 107.4 a | 0.395 a | 4.446 a | |
| V3Ir | 471.5 | 109.4 | 136.8 | 27.4 | 248.4 | 129.8 | 331.7 | 0.277 | 1.913 |
| 279.89 b | |||||||||
| Cr3Ir | 478.6 | 89.6 | 190.2 | 100.4 | 286.3 | 108.5 | 289.0 | 0.332 | 2.639 |
| Nb3Ir | 433.7 | 84.5 | 123.7 | 39.2 | 227.0 | 108.0 | 279.7 | 0.295 | 2.102 |
| 216.4 c | |||||||||
| Mo3Ir | 512.7 | 87.6 | 175.8 | 88.2 | 288.1 | 114.2 | 302.6 | 0.325 | 2.523 |
| 297.5 d | |||||||||
| Compounds | BV | BR | GV | GR | BV/BR | GV/GR | AU |
|---|---|---|---|---|---|---|---|
| Ti3Ir | 171.9 | 171.9 | 35.3 | 17.8 | 1 | 1.984 | 4.922 |
| V3Ir | 248.4 | 248.4 | 132.6 | 127.0 | 1 | 1.044 | 0.220 |
| Cr3Ir | 286.3 | 286.3 | 111.4 | 105.6 | 1 | 1.055 | 0.277 |
| Nb3Ir | 227.0 | 227.0 | 112.7 | 103.3 | 1 | 1.091 | 0.455 |
| Mo3Ir | 288.1 | 288.1 | 119.9 | 108.4 | 1 | 1.106 | 0.532 |
| Crystalline Orientation | Ti3Ir | V3Ir | Cr3Ir | Nb3Ir | Mo3Ir | |
|---|---|---|---|---|---|---|
| [111] | [111]vl | 5226.6 | 6138.7 | 5942.8 | 5460.3 | 5583.6 |
| [11]vt1,2 | 1629.2 | 3762.0 | 3311.6 | 3397.4 | 3301.0 | |
| [110] | [110]vl | 4763.3 | 5856.6 | 5744.8 | 5307.3 | 5466.2 |
| [10]vt1 | 1416.9 | 5656.7 | 5010.3 | 5216.1 | 5093.6 | |
| [001]vt2 | 2440.3 | 3234.2 | 2792.4 | 2723.7 | 2597.2 | |
| [100] | [100]vl | 4551.4 | 6713.4 | 6454.1 | 6169.4 | 6283.3 |
| [010]vt1 | 2440.3 | 3234.2 | 2792.4 | 2723.7 | 2597.2 | |
| [001]vt2 | 2440.3 | 3234.2 | 2792.4 | 2723.7 | 2597.2 | |
| vL | 4833.4 | 6346.8 | 6124.7 | 5706.4 | 5822.9 | |
| vT | 1728.8 | 3522.6 | 3073.2 | 3079.0 | 2965.2 | |
| vD | 1962.7 | 3920.4 | 3444.0 | 3434.1 | 3320.0 | |
| Θ | 233.3 | 487.9 | 441.0 | 396.4 | 397.8 | |
| 238 a, 262.6 b | 460 ± 10 c, 445 d | 449 e | 409 ± 8 c, 377 d | 452 f, 325 g, 497.06 h |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Geng, J.; Wu, Y.; Wang, M.; Xia, C. Insight into Physical and Thermodynamic Properties of X3Ir (X = Ti, V, Cr, Nb and Mo) Compounds Influenced by Refractory Elements: A First-Principles Calculation. Crystals 2019, 9, 104. https://doi.org/10.3390/cryst9020104
Chen D, Geng J, Wu Y, Wang M, Xia C. Insight into Physical and Thermodynamic Properties of X3Ir (X = Ti, V, Cr, Nb and Mo) Compounds Influenced by Refractory Elements: A First-Principles Calculation. Crystals. 2019; 9(2):104. https://doi.org/10.3390/cryst9020104
Chicago/Turabian StyleChen, Dong, Jiwei Geng, Yi Wu, Mingliang Wang, and Cunjuan Xia. 2019. "Insight into Physical and Thermodynamic Properties of X3Ir (X = Ti, V, Cr, Nb and Mo) Compounds Influenced by Refractory Elements: A First-Principles Calculation" Crystals 9, no. 2: 104. https://doi.org/10.3390/cryst9020104
APA StyleChen, D., Geng, J., Wu, Y., Wang, M., & Xia, C. (2019). Insight into Physical and Thermodynamic Properties of X3Ir (X = Ti, V, Cr, Nb and Mo) Compounds Influenced by Refractory Elements: A First-Principles Calculation. Crystals, 9(2), 104. https://doi.org/10.3390/cryst9020104