3.1. Single-Crystal X-ray Diffraction
A single crystal of akdalaite with hexagonal plate morphology was selected from the samples prepared from boehmite (
Supplementary Materials Figure S2). The summary of akdalaite crystallographic data, SC-XRD structural refinement details, refined parameters, and other structural details are provided in
Table A1,
Table A2,
Table A3,
Table A4 and
Table A5 in the
Appendix A. After reducing the SC-XRD data [
26], the crystal structure was solved using a charge flipping algorithm [
27,
28], and inspection of the Wilson plot and the intensity weighted reciprocal lattice, including Bijovet pairs, suggested the structure was acentric with a point group of 6 mm [
29]. We note that the charge flipping algorithm only satisfactorily converged in the noncentrosymmetric space group
P6
3mc. Ab initio structure determination followed by least-squares Fourier cycling confirmed the accepted space group assignment and located the nonhydrogen atoms. Displacement parameters for all the nonhydrogen atoms were refined anisotropically. The hydrogen atom was located from a Fourier difference map and geometrically constrained. The refinement using SC-XRD data confirmed the proposed structure of “tohdite” [
30] and found residual electron density from hydrogen around site O1. The weak X-ray scattering from hydrogen did not allow a full refinement but did provide an adequate starting site position for NPD Rietveld refinements described below. Because the positions of hydrogen, critical for the determination of site bond valence sums, are better determined from the NPD data, we discuss the structural details with reference to the neutron refinement and make reference to the SC-XRD results where they fortify or modify the conclusions derived from the NPD data.
3.3. Solid State NMR
Solid-state NMR data gave additional insight into the local environments of Al and H in akdalaite and confirmed the akdalaite structure solved from SC-XRD and further refined from NPD. The
27Al MAS NMR spectra of synthetic and natural akdalaite (
Figure S4) indicate both samples have six- and four-coordinate Al.
The synthetic sample was further studied with
27Al MQ/MAS to determine the number and proportion of distinct Al sites in akdalaite. The spectra from the
27Al MQ/MAS study revealed three distinct Al sites—two octahedral and one tetrahedral (Figure 4)—with isotropic peaks at 11.5, 23.4, and 77.5 ppm in F1 (vertical axis in
Figure 3). Simulation of the separate F2 cross sections to second-order quadrupolar peak shapes (
Figure 3, right panel) yielded the chemical shift (δiso), quadrupolar coupling constant (Cq), and asymmetry parameter (η) for each resolved site, compiled in
Table 1. These values were then used to fit the quantitative MAS spectrum to a sum of second-order quadrupolar center bands, yielding an Al site distribution of 59% ± 2% for the Al1 octahedral site, 22% ± 2% for the Al2 octahedral site, and 20% ± 2% for the Al3 tetrahedral site, in reasonable agreement with the X-ray- and neutron-derived models.
The
1H NMR spectrum consists primarily of a single, narrow central peak at +8.6 ppm, with two spinning sidebands, as shown in
Figure 4. A small shoulder near +6.5 ppm (<5% intensity) likely arises from an impurity phase and/or surface adsorbed water. The single, well-resolved central peak indicates a single H site, and the small peak width (600 Hz) observed at this modest spinning rate (15 kHz) indicates a small homonuclear dipolar coupling characteristic of low hydrogen density [
35,
36]. In this chemical shift range, the literature data for
1H indicate a good linear correlation with d(O…O) hydrogen bond lengths [
35], although the relationship between chemical shift and hydrogen bond distance can also be expected to reflect other geometrical factors such as H–O distance and O–H…O angle. With that caveat, the observed shift at 8.6 ppm (
Figure 4) is similar to that of diaspore (α-AlOOH), which has a shift at +9.4 ppm and an O···O distance of ~2.65 Å [
35] compared with an O…O distance of ~2.91 Å in akdalaite.
3.4. Crystal Chemical Analysis
The results from SC-XRD, NPD, and NMR were used to form a complete structural model of akdalaite (
Figure 5 and
Figure 6) for comparison with ferrihydrite [
9]. The bond valence sums (BVS) were calculated from the NPD data in the same manner as ferrihydrite, where the total site valence (
) is the sum of valences (
) associated with the bond between atoms
i and
j [
37,
38,
39]. The individual bond valences were calculated using Equation (1) with the experimentally determined bond lengths (
Rij) and the empirical constants
R0 and b determined from valence analysis of known compounds. For Al–O bonds,
R0 = 1.651 and b = 0.37 [
19,
37].
The interatomic distances and bond valence sum listed in
Table A8 and
Table A9, and overlaid on the structure in
Figure 5 and
Figure 6, broadly conform to values expected from the isostructural MM model of ferrihydrite [
9]. The contribution of hydrogen to the oxygen bond valence was accomplished using different empirical constants for the shorter donor O–H and longer acceptor O…H bonds (
Table A8). Previous studies found that hydrogen bonding is unlike typical cation–anion interactions and the hydrogen bonding interactions have to be treated differently [
38,
39,
40]. The aluminum sites show signs of over- and underbonding as the BVS of the tetrahedral site (Al3) is underbonded at 2.71 Å, below the ideal value of 3 for aluminum (
Figure 6) but conforming to the result reported for the MM model (
Figure 1). The octahedral aluminum sites of Al1 and Al2 are closer to ideal being, 3.11 and 2.79 Å, respectively. This is similar to Fe BVS of the ferrihydrite model (
Figure 1), where the octahedral sites are 2.97 while the tetrahedral site is underbonded at 2.74.
The oxygen BVS sums are also similar to those found in ferrihydrite, with the O4 site overbonded at 2.15 and 2.29 in akdalaite (
Figure 6) and ferrihydrite (
Figure 1), respectively. The hydroxyl oxygen (O1) has an understandably low BVS at 1.44 prior to accounting for hydrogen bonding. After accounting for hydrogen bonding, the oxygen sites are slightly over- and underbonded (
Table A4 and
Table A9). The akdalaite model, especially that derived from the neutron data, correctly takes account of the H contribution to the BVS for oxygen. In the case of ferrihydrite, the oxygen BVS is estimated. With this caveat, the akdalaite and ferrihydrite BVS are quite comparable, further suggesting deviations from ideality are inherent to bond strain associated with the structure type.
In addition, for any nonideality in bond valence sum, some of the Al bonding environments in akdalaite have characteristics that are similar to those observed in the ferrihydrite model. In analyzing the Al–Al distances (
Table A10,
Figure 5), it is clear they are nonideal. The Al1–Al1 bond distance between the edge sharing octahedra is 2.802 Å, much shorter than the other edge sharing octahedra where the Al1–Al2 distance is 3.018 Å (
Table A10). The short edge sharing distance between Al1–Al1 is close to that of the Al octahedral face sharing distance of corundum at 2.654 Å and is ~9% shorter than the other edge sharing Al1–Al2 distance, 3.339 Å (
Table A4,
Figure 5). In ferrihydrite, the Fe1–Fe1 bond distance is 2.907 Å, which is ~11% shorter than the other shared edge Fe1–Fe2 bond distance of 3.261 Å, much like the Al sites in akdalaite. However, this Fe1–Fe1 edge sharing distance is only slightly larger than the Fe octahedra face sharing distance in hematite (2.895 Å) [
20,
41].
The Al2 octahedra in akdalaite are also distorted in that the Al is off center so much that three of the Al2–O distances are only 1.874(3) Å, which are close to the Al–O bond distances in the Al3-centered tetrahedron, between 1.788(1) and 1.818(4) Å (
Figure 5,
Table A8). The ferrihydrite Fe2 octahedral distortion also results in uncharacteristically short Fe–O distances of 1.881 Å, which in turn are shorter than some of the tetrahedral Fe–O distances (1.931 Å). The atypical bonding at the Al2 site observed in akdalaite is also present at the Fe2 site in the ferrihydrite model, again suggesting considerably strained structures, to the point of violating accepted principles [
21].
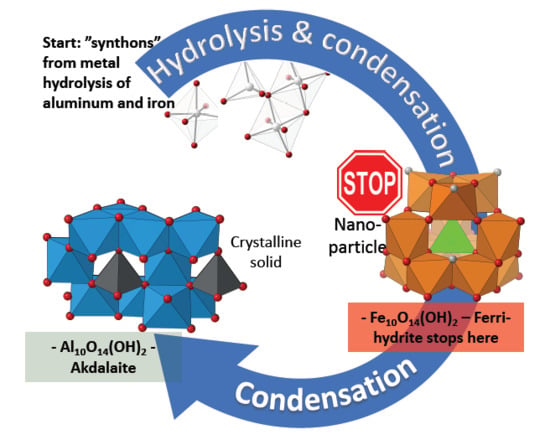
Thorough crystal chemical analysis has revealed that most of the anomalous aspects of the ferrihydrite model are intrinsic to the akdalaite structure. The only structural oddity of the MM ferrihydrite model [
9] not observed in the akdalaite structure, and for which the MM model was criticized, is the distorted edge sharing O–O bonding distances between the Fe1 sites where the shared edge was longer than the unshared edge (
Figure 1). The Al1 octahedra have a slightly shorter O–O distance on the shared edge (2.617 Å) in comparison with the unshared edge (2.697 Å) in accordance with Pauling’s distortion rule [
42]. Otherwise, the anomalous crystal chemical trends that are observed in akdalaite are present in the ferrihydrite model. These differences have been discussed previously as possibly arising from the assumptions inherent in the fitting of PDF data from which the MM model is derived.
It was emphasized in the original single-phase ferrihydrite model refinement that while the structure can be described by an idealized periodic model, real-space fitting does not take into account that “second-order effects such as disorder, surface relaxation, internal strain, defects (e.g., stacking faults), particle shape, and/or interparticle correlations may also contribute to the experimental PDFs” [
10]. The PDF patterns of nanoparticles are heavily influenced by the size and morphology of the particles, as this will weight some pair correlations more than others [
43,
44,
45]. Treatment of the nanocrystal morphology, such as modeling the ferrihydrite plate morphology as a sphere, can result in model distortions in order to better fit the data.
Further, as the nanoparticles of ferrihydrite have a high surface area, surface interaction and relaxation will also have a significant impact on the total scattering. In crystals and crystallites, the surface-area-to-volume ratio is so small that surface structure will have a negligible contribution to the scattering. However, in the case of nanoparticles such as ferrihydrite, the surface comprises a significant portion of the structure and surface contribution to the total scattering is no longer minor. A study of ferrihydrite surface structure elucidated that the Fe sites exposed at the surface are dominantly the Fe1 octahedra [
46]. The shorter-than-expected Fe1–Fe1 distance and distortion of the Fe1 octahedra is likely due to the strains placed on the sites at the surface of ferrihydrite particles. Thus, surface interactions and relaxation will preferentially distort the Fe1 octahedra and the distortions on the surface will have a significant contribution to the total scattering and PDF. Thus, when a unit cell approach to modeling the total scattering and PDF is used, the bulk Fe1 site will appear distorted due to the contributions of distorted Fe1 octahedra at the surface—the bulk and surface Fe1 sites are average in such an approach. While the previous critiques of the ferrihydrite crystal chemistry were originally valid, crystal chemical analysis has shown that these are inherent characteristics of the akdalaite model, and exacerbated distortions likely arise from the nonideal treatment of PDF data during model refinement. It is for this reason that the fully ordered structure of akdalaite is an important benchmark for the isostructural bulk structure of ferrihydrite and an excellent starting point for calculations of the distortions resulting from disorder, partial site occupancy, and surface relaxation. Further validation of the appropriateness of the akdalaite model as a proxy for the calculation of possible ferrihydrite disordered structures is provided in the
Supplementary Materials.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






