Novel Cerium Bisphosphinate Coordination Polymer and Unconventional Metal–Organic Framework

 ,
,
Abstract
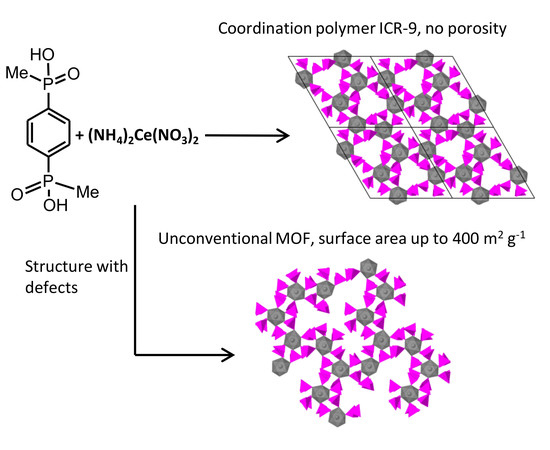
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of ICR-9
2.2.1. Synthesis of Well-Crystalline ICR-9
2.2.2. Synthesis of ICR-9 with UMOF Phase
2.3. Instrumental Methods
3. Results and Discussion
3.1. Synthesis
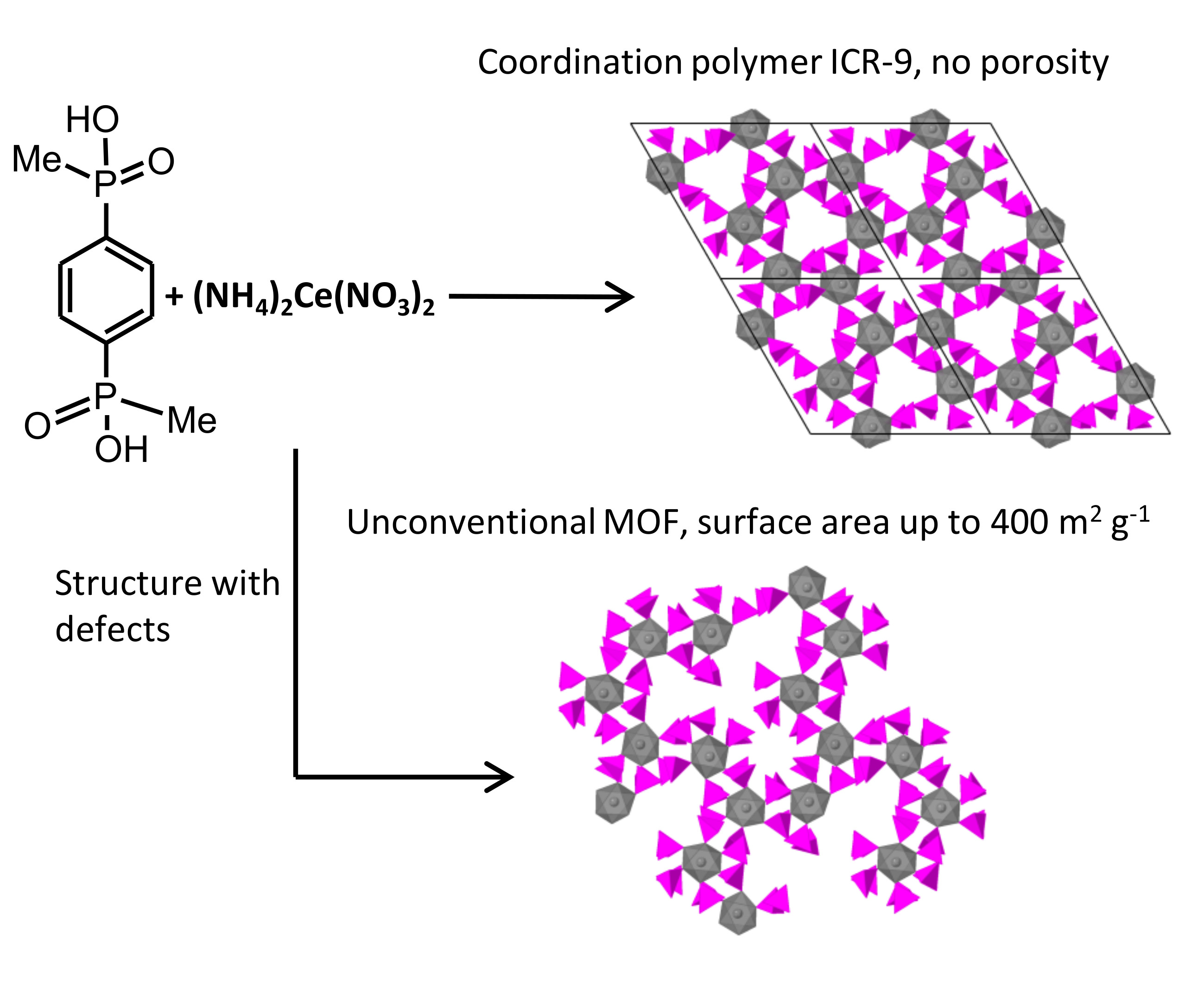
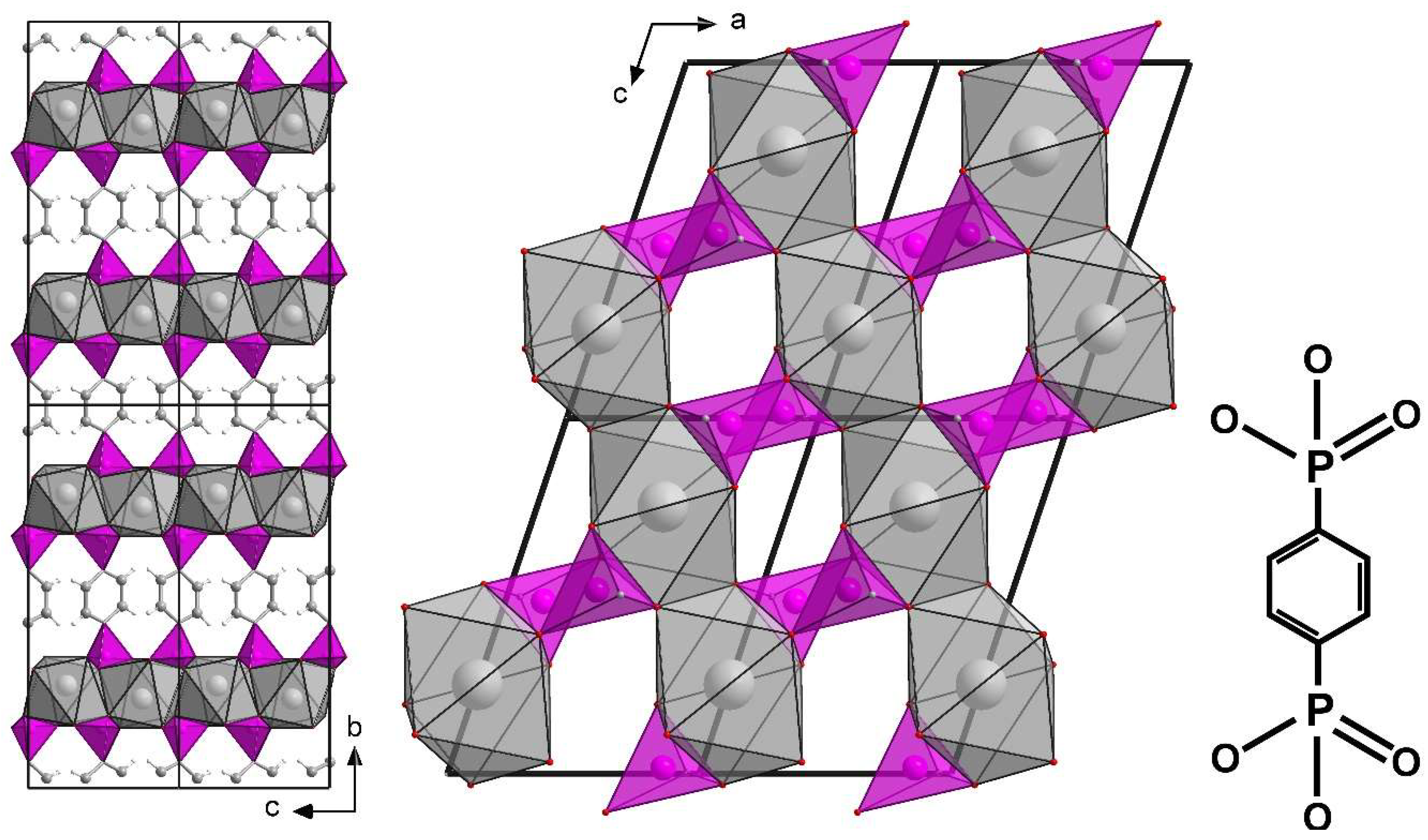
3.2. Structure of ICR-9
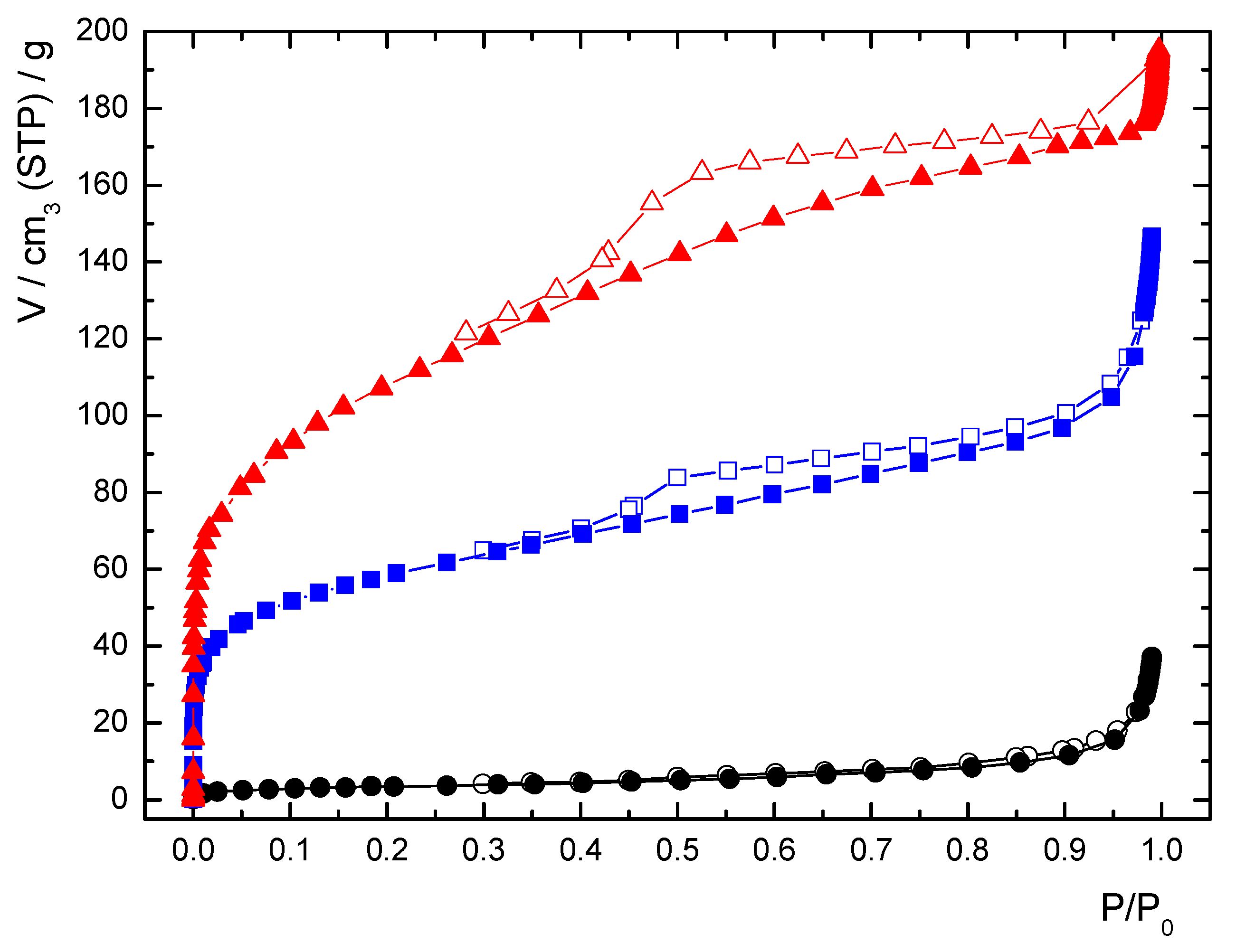
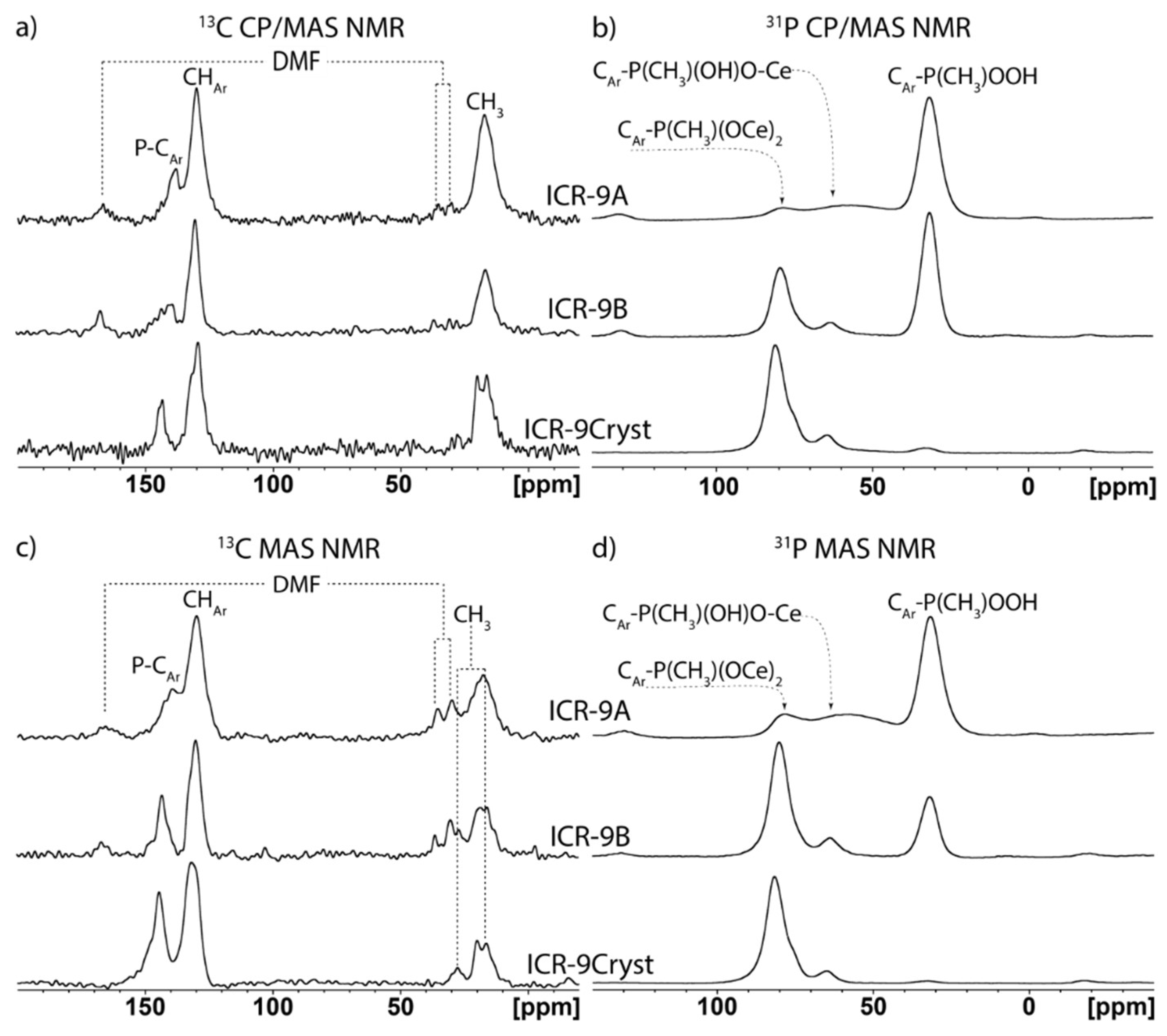
3.3. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaskel, S. The Chemistry of Metal–Organic Frameworks: Synthesis, Characterization, and Applications; Wiley-VCH: Weinheim, Germany, 2017; Volume 2. [Google Scholar]
- Leus, K.; Bogaerts, T.; De Decker, J.; Depauw, H.; Hendrickx, K.; Vrielinck, H.; Van Speybroeck, V.; Van Der Voort, P. Systematic study of the chemical and hydrothermal stability of selected “stable” Metal Organic Frameworks. Microporous Mesoporous Mater. 2016, 226, 110–116. [Google Scholar] [CrossRef]
- Bůžek, D.; Demel, J.; Lang, K. Zirconium Metal-Organic Framework UiO-66: Stability in Aqueous Environment and its Relevance for Organophosphate Degradations. Inorg. Chem. 2018, 57, 14290–14297. [Google Scholar] [CrossRef] [PubMed]
- Bůžek, D.; Zelenka, J.; Ulbrich, P.; Ruml, T.; Křížová, I.; Lang, J.; Kubát, P.; Demel, J.; Kirakci, K.; Lang, K. Nanoscaled porphyrinic metal-organic frameworks: Photosensitizer delivery systems for photodynamic therapy, J. Mater. Chem. B 2017, 5, 1815–1821. [Google Scholar] [CrossRef]
- Clearfield, A.; Demadis, K. Metal Phosphonate Chemistry: From Synthesis to Applications; Royal Society of Chemistry: Oxford, UK, 2012. [Google Scholar]
- Taddei, M.; Costantino, F.; Vivani, R. Robust Metal-Organic Frameworks Based on Tritopic Phosphonoaromatic Ligands. Eur. J. Inorg. Chem. 2016, 2016, 4300–4309. [Google Scholar] [CrossRef]
- Shearan, S.J.I.; Stock, S.; Emmerling, F.; Demel, J.; Wright, P.A.; Demadis, K.D.; Vassaki, M.; Costantino, F.; Vivani, R.; Sallard, S.; et al. New Directions in Metal Phosphonate and Phosphinate Chemistry. Crystals 2019, 9, 270. [Google Scholar] [CrossRef]
- Gagnon, K.J.; Perry, H.P.; Clearfield, A. Conventional and Unconventional Metal-Organic Frameworks Based on Phosphonate Ligands: MOFs and UMOFs. Chem. Rev. 2012, 112, 1034–1054. [Google Scholar] [CrossRef] [PubMed]
- Carson, I.; Healy, M.R.; Doidge, E.D.; Love, J.B.; Morrison, C.A.; Tasker, P.A. Metal-binding motifs of alkyl and aryl phosphinates; versatile mono and polynucleating ligands. Coord. Chem. Rev. 2017, 335, 150–171. [Google Scholar] [CrossRef] [Green Version]
- Du, J.-L.; Rettig, S.J.; Thompson, R.C.; Trotter, J. Synthesis, structure, and magnetic properties of diphenylphosphinates of cobalt(II) and manganese(II). The crystal and molecular structures of the γ forms of poly-bis(μ-diphenylphosphinato)cobalt(II) and manganese(II). Can. J. Chem. 1991, 69, 277–285. [Google Scholar] [CrossRef]
- Rosca, I.; Nechita, M.-T.; Sutiman, D.; Cailean, A.; Sibiescu, D.; Vizitiu, M. New iron (III) coordination compounds with applications in water treatment. Environ. Eng. Manag. J. 2010, 9, 511–517. [Google Scholar]
- Oleshkevich, E.; Viñas, C.; Romero, I.; Choquesillo-Lazarte, D.; Haukka, M.; Teixidor, F. M-Carboranylphosphinate as Versatile Building Blocks to Design all Inorganic Coordination Polymers. Inorg. Chem. 2017, 56, 5502–5505. [Google Scholar] [CrossRef]
- Oleshkevich, E.; Teixidor, F.; Rosell, A.; Viñas, C. Merging Icosahedral Boron Clusters and Magnetic Nanoparticles: Aiming toward Multifunctional Nanohybrid Materials. Inorg. Chem. 2018, 57, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.-Y.; Zhang, L.; Wang, B.-Y.; Liu, S.-J.; Huang, B.; Liu, C.-M.; Zhang, W.-X. Two magnetic Δ-chain-based Mn(II) and Co(II) coordination polymers with mixed carboxylate-phosphinate and μ3-OH– bridges. CrystEngComm. 2017, 19, 1052–1057. [Google Scholar] [CrossRef]
- Yang, W.; Wang, H.; Tian, W.-G.; Li, J.; Sun, Z.-M. The first family of actinide carboxyphosphinates: Two- and three-dimensional uranyl coordination polymers. Eur. J. Inorg. Chem. 2014, 31, 5378–5384. [Google Scholar] [CrossRef]
- Li, J.; Xue, C.-C.; Liu, S.; Wang, Z.-X. Structures and magnetic properties of two noncentrosymmetric coordination polymers based on carboxyphosphinate ligand. Solid State Sci. 2016, 61, 111–115. [Google Scholar] [CrossRef]
- Cecconi, F.; Dakternieks, D.; Duthie, A.; Ghilardi, C.A.; Gili, P.; Lorenzo-Luis, P.A.; Midollini, S.; Orlandini, A. Inorganic-organic hybrids of the p,p′-diphenylmethylenediphosphinate ligand with bivalent metals: A new 2D-layered phenylphosphinate zinc(II) complex. J. Solid State Chem. 2004, 177, 786–792. [Google Scholar] [CrossRef]
- Shekurov, R.; Miluykov, V.; Kataeva, O.; Krivolapov, D.; Sinyashin, O.; Gerasimova, T.; Katsyuba, S.; Kovalenko, V.; Krupskaya, Y.; Kataev, V.; et al. Inorganic-organic hybrids of the p,p′-diphenylmethylenediphosphinate ligand with bivalent metals: A new 2D-layered phenylphosphinate zinc(II) complex. Cryst. Growth Des. 2016, 16, 5084–5090. [Google Scholar] [CrossRef]
- Shekurov, R.; Khrizanforova, V.; Gilmanova, L.; Khrizanforov, M.; Miluykov, V.; Kataeva, O.; Yamaleeva, Z.; Burganov, T.; Gerasimova, T.; Khamatgalimov, A.; et al. Zn and Co redox active coordination polymers as efficient electrocatalysts. Dalton Trans. 2019, 48, 3601–3609. [Google Scholar] [CrossRef] [PubMed]
- Hynek, J.; Brázda, P.; Rohlíček, J.; Londesborough, M.G.S.; Demel, J. Phosphinic Acid Based Linkers: Building Blocks in Metal–Organic Framework Chemistry. Angew. Chem. Int. Ed. 2018, 130, 5016–5019. [Google Scholar] [CrossRef]
- Costantino, F.; Gentili, P.L.; Audebrand, N. A new dual luminescent pillared cerium(IV)sulfate–diphosphonate. Inorg. Chem. Commun. 2009, 12, 406–408. [Google Scholar] [CrossRef]
- Lammert, M.; Wharmby, M.T.; Smolders, S.; Bueken, B.; Lieb, A.; Lomachenko, K.A.; De Vos, D.; Stock, N. Cerium-based metal organic frameworks with UiO-66 architecture: Synthesis, properties and redox catalytic activity. Chem. Commun. 2015, 51, 12578–12581. [Google Scholar] [CrossRef]
- Lammert, M.; Reinsch, H.; Murray, C.A.; Wharmby, M.T.; Terraschke, H.; Stock, N. Synthesis and Structure of Zr(IV)- and Ce(IV)-Based CAU-24 with 1,2,4,5-Tetrakis(4-Carboxyphenyl)-Benzene. Dalton Trans. 2016, 45, 18822–18826. [Google Scholar] [CrossRef] [PubMed]
- Almáši, M.; Zeleňák, V.; Opanasenko, M.; Císařová, I. Ce(III) and Lu(III) Metal-Organic Frameworks with Lewis Acid Metal Sites: Preparation, Sorption Properties and Catalytic Activity in Knoevenagel Condensation. Catal. Today 2015, 243, 3098–3114. [Google Scholar] [CrossRef]
- Atzori, C.; Lomachenko, K.A.; Øien-Ødegaard, S.; Lamberti, C.; Stock, N.; Barolo, C.; Bonino, F. Disclosing the Properties of a New Ce(III)-Based MOF: Ce2(NDC)3(DMF)2 Cryst. Growth Des. 2019, 19, 787–796. [Google Scholar] [CrossRef]
- Vincent, R.; Midgley, P.A. Double conical beam-rocking system for measurement of integrated electron diffraction intensities. Ultramicroscopy 1994, 53, 271–282. [Google Scholar] [CrossRef]
- Palatinus, L. PETS–Program for Analysis of Electron Diffraction Data; Institute of Physics of the Czech Academy of Sciences: Prague, Czech Republic, 2011. [Google Scholar]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Kristallogr. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Equbal, C.A.; Bjerring, M.; Madhu, P.K.; Nielsen, N.C. Improving spectral resolution in biological solid-state NMR using phase-alternated rCW heteronuclear decoupling. Chem. Phys. Lett. 2015, 635, 339–344. [Google Scholar] [CrossRef]
- Brus, J. Heating of Samples induced by fast magic-angle spinning. Solid State Nucl. Magn. Reson. 2000, 16, 151–160. [Google Scholar] [CrossRef]
- Rhauderwiek, T.; Heidenreich, N.; Reinsch, H.; Øien-Ødegaard, S.; Lomachenko, K.A.; Rütt, U.; Soldatov, A.V.; Lillerud, K.P.; Stock, N. Co-Ligand Dependent Formation and Phase Transformation of Four Porphyrin-Based Cerium Metal–Organic Frameworks. Cryst. Growth Des. 2017, 17, 3462–3474. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Cryst. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- CCDC no. 1913129. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 30 April 2019).
- Amghouz, Z.; García-Granda, S.; García, J.R.; Clearfield, A.; Valiente, R. Organic-Inorganic Hybrids Assembled from Lanthanide and 1,4-Phenylenebis(phosphonate). Cryst. Growth Des. 2011, 11, 5289–5297. [Google Scholar] [CrossRef]
- Bennett, T.D.; Cheetham, A.K. Amorphous Metal−Organic Frameworks. Acc. Chem. Res. 2014, 47, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Shearer, G.C.; Chavan, S.; Bordiga, S.; Svelle, S.; Olsbye, U.; Lillerud, K.P. Defect Engineering: Tuning the Porosity and Composition of the Metal−Organic Framework UiO-66 via Modulated Synthesis. Chem. Mater. 2016, 28, 3749–3761. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | mol% DMFa* | mol% of Uncoordinated PBP(Me)2− b* | Specific Surface Area / m2 g−1 | Estimation of the Amorphous Content / wt%c** |
|---|---|---|---|---|
| ICR-9Cryst | <5% | 1% | 13 | 0% |
| ICR-9B | 37% | 21% | 211 | 36% |
| ICR-9A | 31% | 61% | 395 | 77% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohlíček, J.; Bůžek, D.; Brázda, P.; Kobera, L.; Hynek, J.; Brus, J.; Lang, K.; Demel, J. Novel Cerium Bisphosphinate Coordination Polymer and Unconventional Metal–Organic Framework. Crystals 2019, 9, 303. https://doi.org/10.3390/cryst9060303
Rohlíček J, Bůžek D, Brázda P, Kobera L, Hynek J, Brus J, Lang K, Demel J. Novel Cerium Bisphosphinate Coordination Polymer and Unconventional Metal–Organic Framework. Crystals. 2019; 9(6):303. https://doi.org/10.3390/cryst9060303
Chicago/Turabian StyleRohlíček, Jan, Daniel Bůžek, Petr Brázda, Libor Kobera, Jan Hynek, Jiří Brus, Kamil Lang, and Jan Demel. 2019. "Novel Cerium Bisphosphinate Coordination Polymer and Unconventional Metal–Organic Framework" Crystals 9, no. 6: 303. https://doi.org/10.3390/cryst9060303
APA StyleRohlíček, J., Bůžek, D., Brázda, P., Kobera, L., Hynek, J., Brus, J., Lang, K., & Demel, J. (2019). Novel Cerium Bisphosphinate Coordination Polymer and Unconventional Metal–Organic Framework. Crystals, 9(6), 303. https://doi.org/10.3390/cryst9060303