Platonic Relationships in Metal Phosphonate Chemistry: Ionic Metal Phosphonates


 , ,
, ,
Abstract
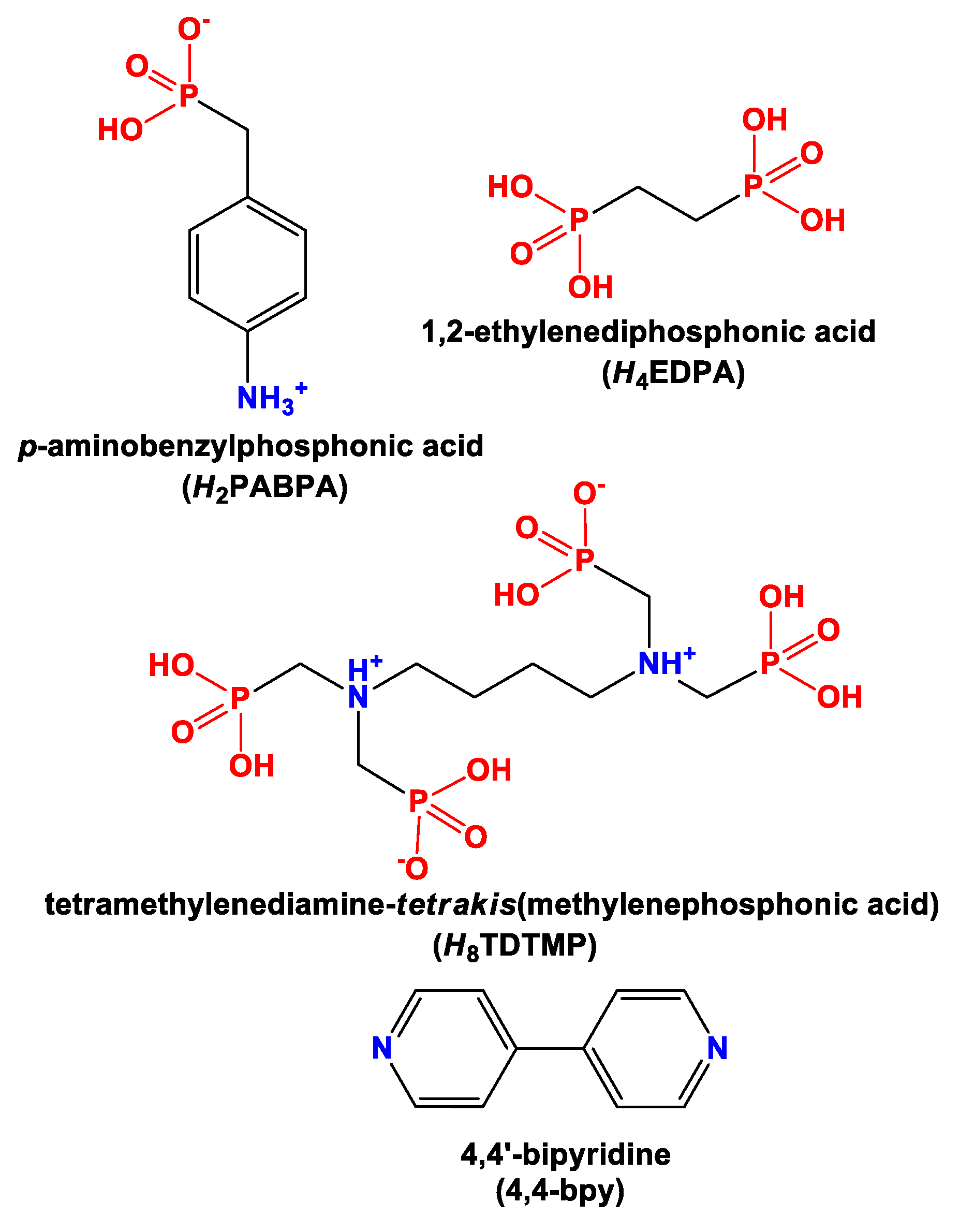
:
1. Introduction
2. Materials and Methods
2.1. Instrumentation
2.2. General
2.3. Synthesis of 4-(bromomethyl)nitrobenzene
2.4. Synthesis of 4-(diethoxyphosphorylmethyl)nitrobenzene
2.5. Synthesis of 4-(diethoxyphosphorylmethyl)aniline
2.6. Synthesis of 4-(dihydroxyphosphorylmethyl)aniline (H2PABPA)
2.7. Synthesis of [Mg(H2O)6]·[HPABPA]2·6H2O (Mg-PABPA)
2.8. Synthesis of [Ca(H2O)8]·[HPABPA]2 (Ca-PABPA)
2.9. Synthesis of [Sr(H2O)8]·[HPABPA]2 (Sr-PABPA)
2.10. Synthesis of [Ni(H2O)6]·[H2EDPA]·H2O (Ni-EDPA)
2.11. Synthesis of {[Ni(4,4’-bpy)(H2O)4]·[H2EDPA]·H2O}n (Ni-bpy-EDPA)
2.12. Synthesis of [Mg(H2O)6]·[H6TDTMP] (Mg-TDTMP)
2.13. Crystal Data Collection And Refinement
2.14. Computational Studies
3. Results
3.1. Synthetic Considerations
3.2. Materials Characterization
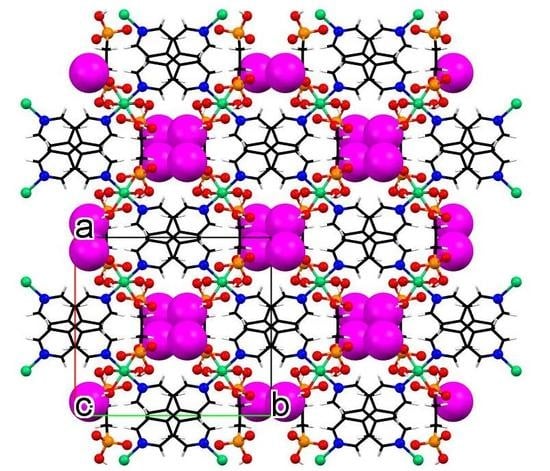
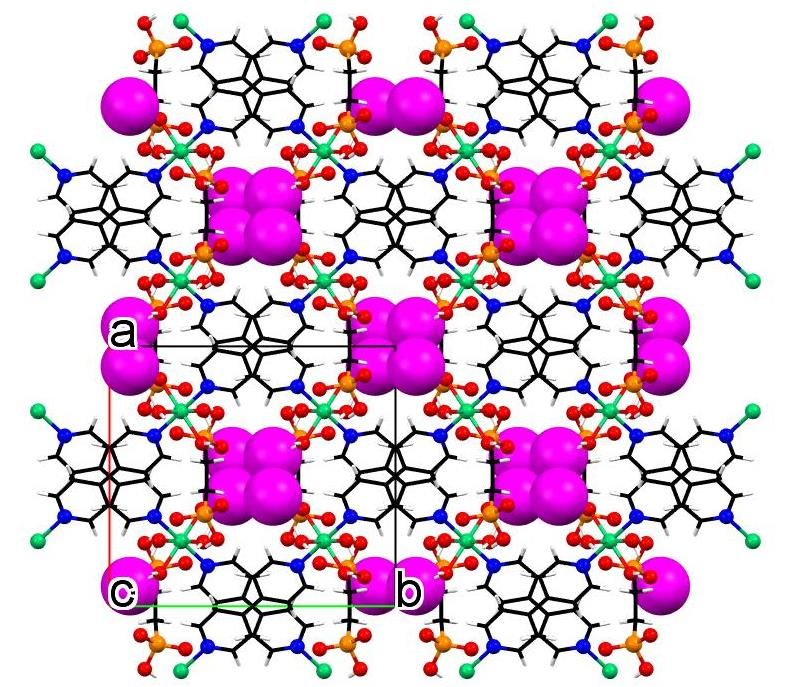
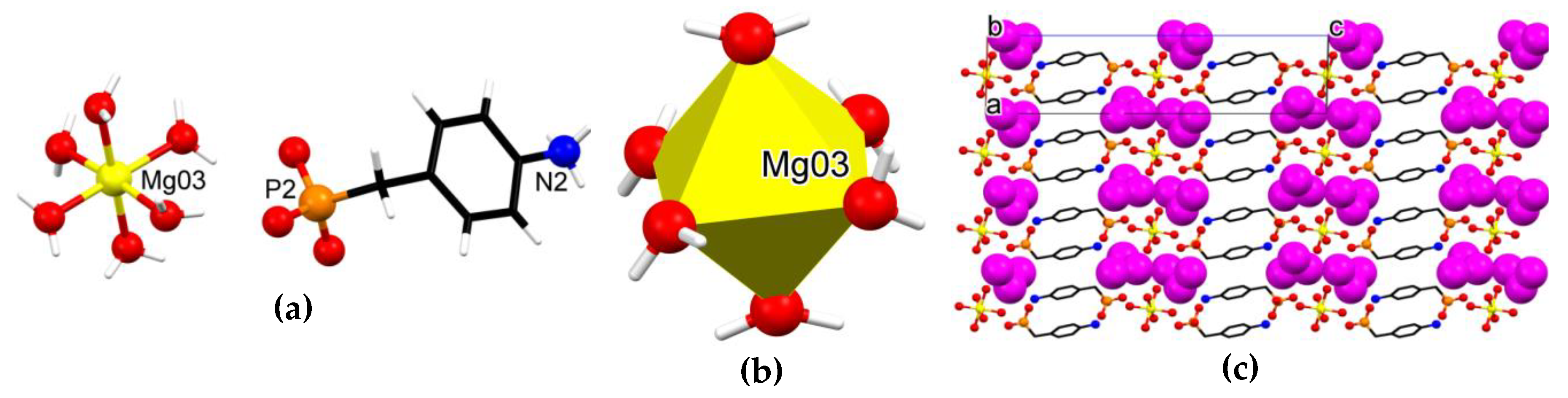
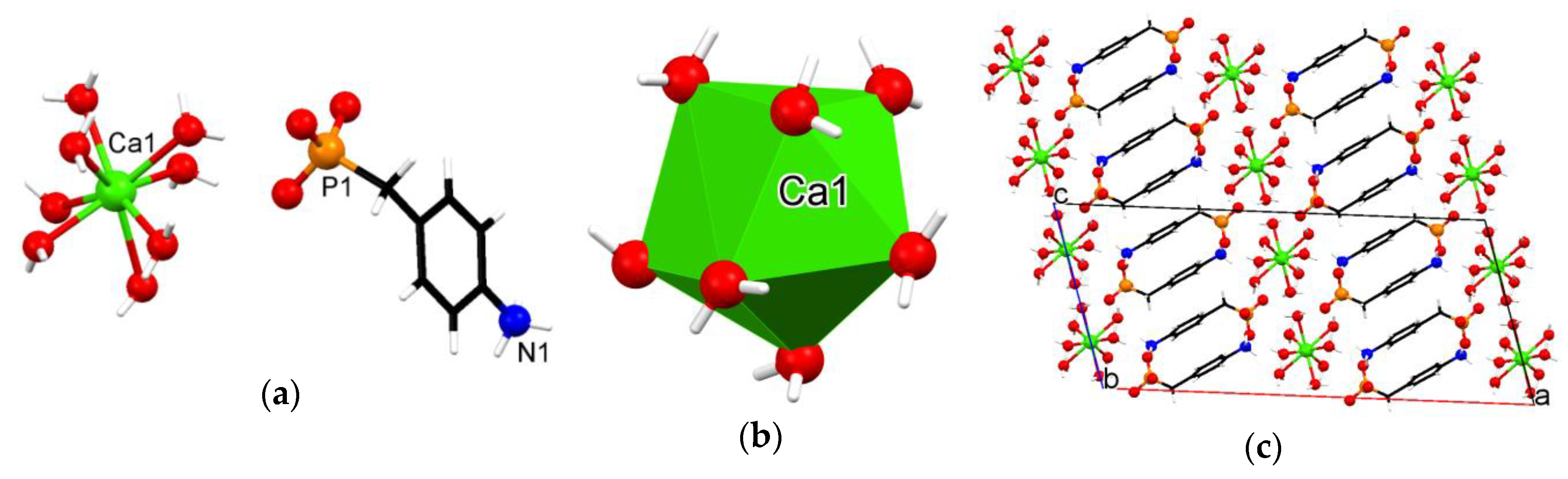
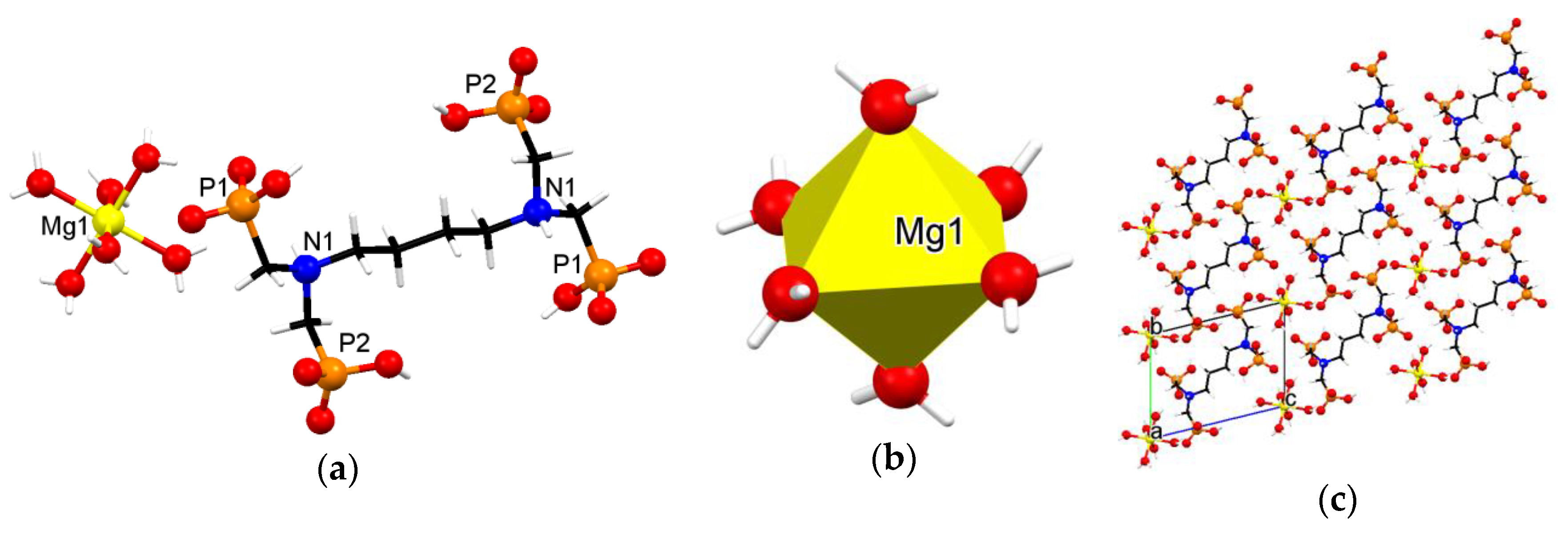
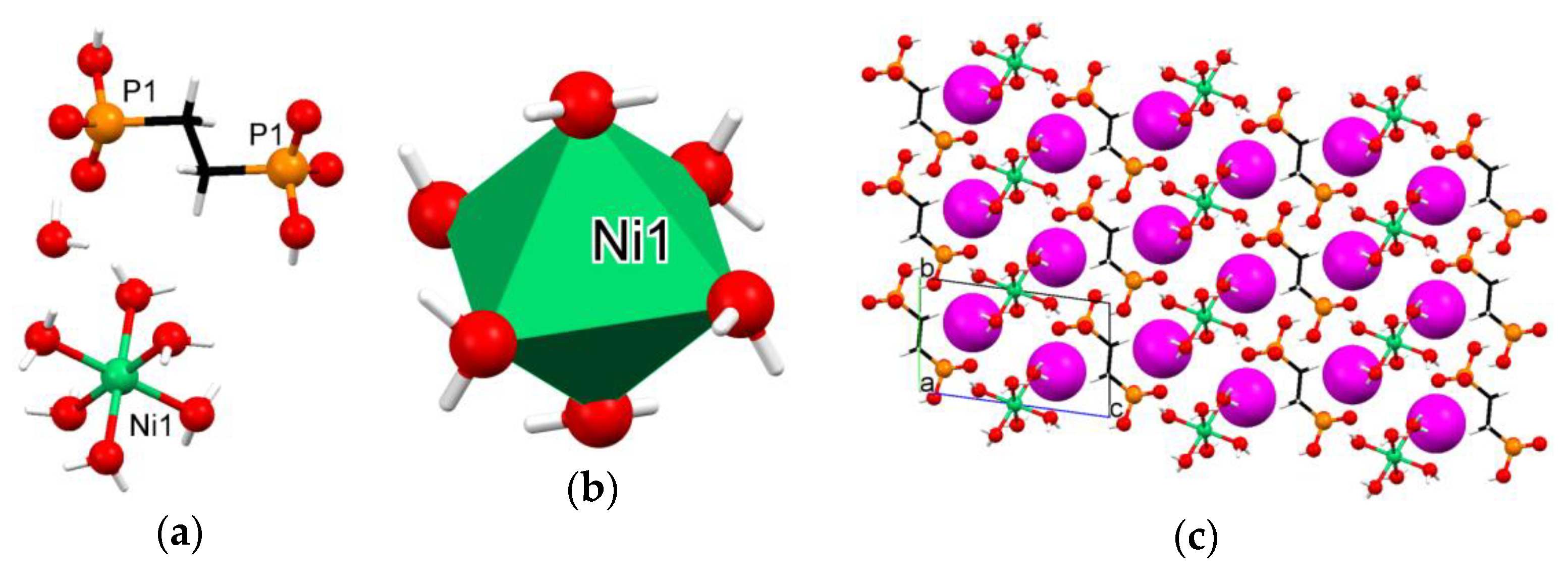
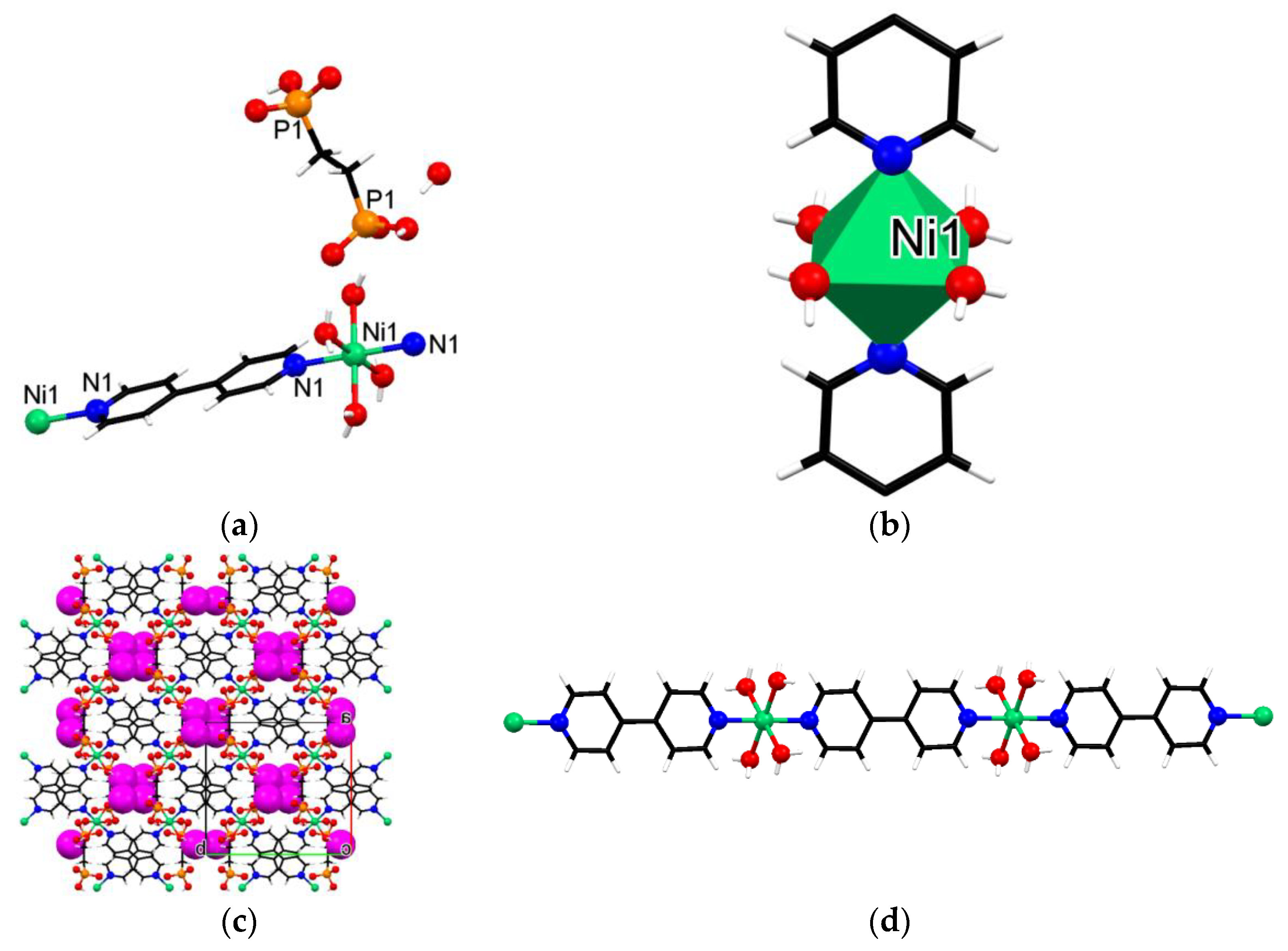
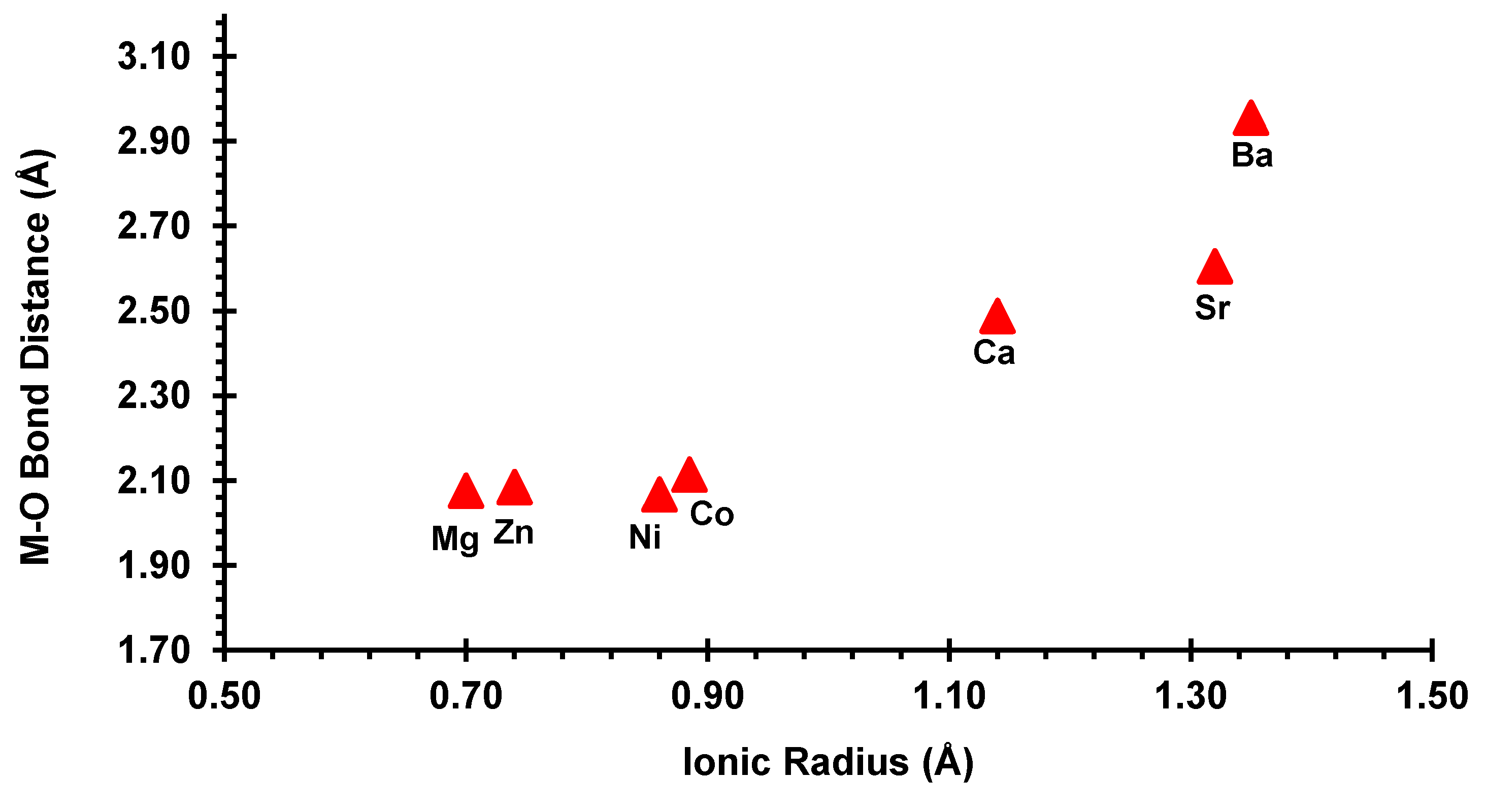
3.3. Crystallographic Description
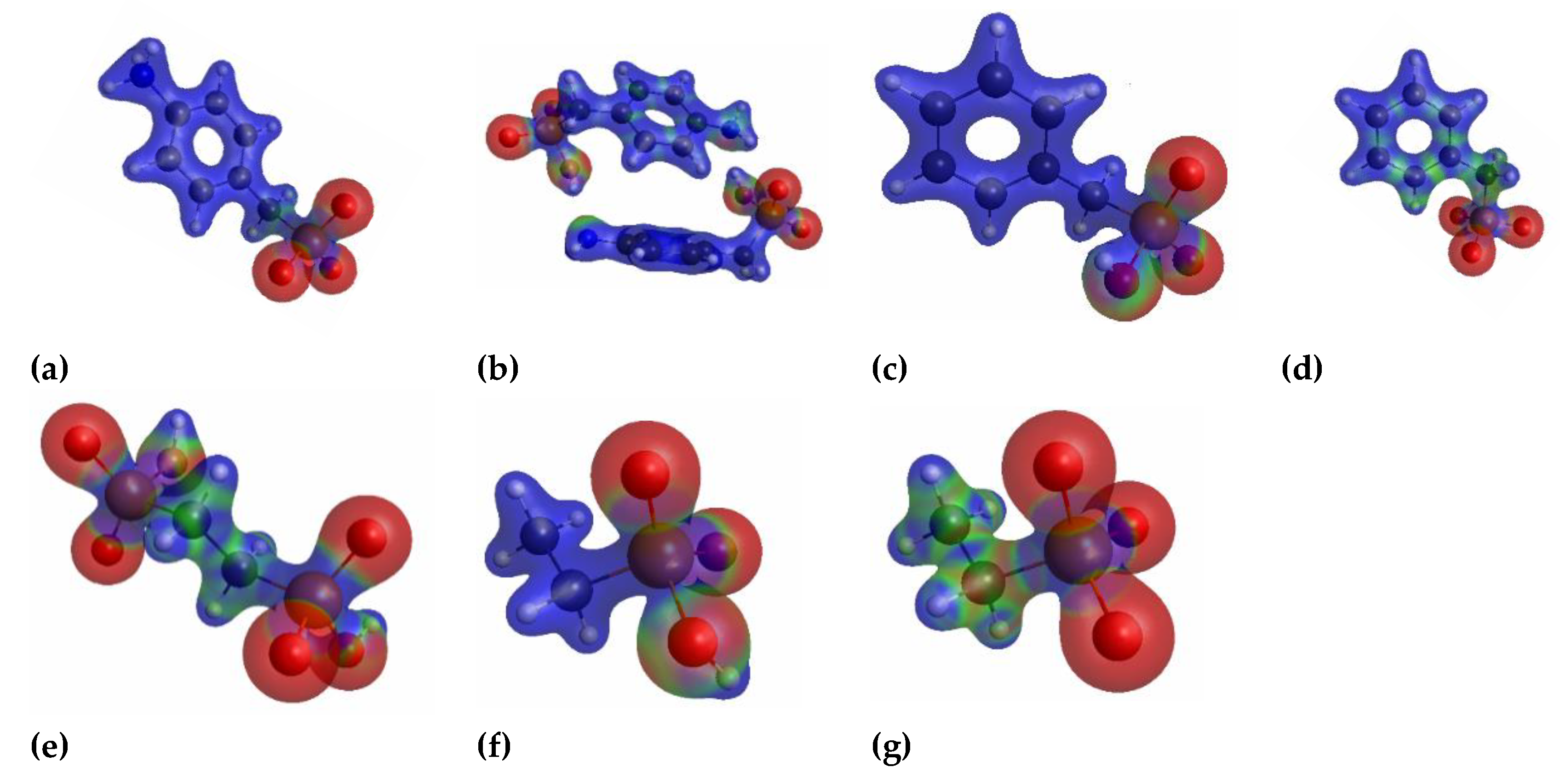
3.4. Computational Results: Total Electronic Densities and Partial Charges
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clearfield, A.; Demadis, K.D. Metal Phosphonate Chemistry: From Synthesis to Applications; Royal Society of Chemistry: London, UK, 2012. [Google Scholar]
- Demadis, K.D.; Stavgianoudaki, N. Structural diversity in metal phosphonate frameworks: Impact on applications. In Metal Phosphonate Chemistry: From Synthesis to Applications; Clearfield, A., Demadis, K.D., Eds.; Royal Society of Chemistry: London, UK, 2012; Chapter 14; pp. 438–492. [Google Scholar]
- Zaręba, J.K. Tetraphenylmethane and tetraphenylsilane as building units of coordination polymers and supramolecular networks–A focus on tetraphosphonates. Inorg. Chem. Commun. 2017, 86, 172–186. [Google Scholar] [CrossRef]
- Bao, S.-S.; Shimizu, G.K.H.; Zheng, L.-M. Proton conductive metal phosphonate frameworks. Coord. Chem. Rev. 2019, 378, 577–594. [Google Scholar] [CrossRef]
- Groves, J.A.; Miller, S.R.; Warrender, S.J.; Mellot-Draznieks, C.; Lightfoot, P.; Wright, P.A. The first route to large pore metal phosphonates. Chem. Commun. 2006, 31, 3305–3307. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, K.E.; Vassaki, M.; Spinthaki, A.; Alatzoglou, F.-E.G.; Tripodianos, E.; Turhanen, P.; Demadis, K.D. Phosphorus chemistry: From small molecules to polymers to pharmaceutical and industrial applications. Pure Appl. Chem. 2019, 91, 421–441. [Google Scholar] [CrossRef]
- Shah, B.; Chudasama, U. Application of zirconium phosphonate—a novel hybrid material as an ion exchanger. Des. Wat. Treat. 2012, 38, 227–235. [Google Scholar]
- Armakola, E.; Colodrero, R.M.P.; Bazaga-García, M.; Salcedo, I.R.; Choquesillo-Lazarte, D.; Cabeza, A.; Kirillova, M.V.; Kirillov, A.M.; Demadis, K.D. Three-component copper-phosphonate-auxiliary ligand systems: Proton conductors and efficient catalysts in mild oxidative functionalization of cycloalkanes. Inorg. Chem. 2018, 57, 10656–10666. [Google Scholar] [CrossRef] [PubMed]
- Moschona, A.; Plesu, N.; Mezei, G.; Thomas, A.; Demadis, K.D. Corrosion protection of carbon steel by tetraphosphonates of systematically different molecular size. Corr. Sci. 2018, 145, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Gałezowska, J.; Kafarski, P.; Kozłowski, H.; Młynarz, P.; Nurchi, V.M.; Pivetta, T. N,N’-ethylenediaminobis(benzylphosphonic acids) as a potent class of chelators for metal ions. Inorg. Chim. Acta 2009, 362, 707–713. [Google Scholar] [CrossRef]
- Heering, C.; Nateghi, B.; Janiak, C. Charge-assisted hydrogen-bonded networks of NH4+ and [Co(NH3)6]3+ with the new linker anion of 4-phosphono-biphenyl-4’-carboxylic acid. Crystals 2016, 6, 22. [Google Scholar] [CrossRef]
- Wilk, M.; Janczak, J.; Videnova-Adrabinska, V. Hexaaquacobalt(II) bis[hydrogen bis(4-carboxyphenylphosphonate)] dihydrate. Acta Crystallogr. Sect. C—Cryst. Struct. Commun. 2011, 67, 9–12. [Google Scholar] [CrossRef]
- Gholivand, K.; Farrokhi, A.R. Supramolecular hydrogen-bonded frameworks from a new bisphosphonic Acid and transition metal ions. Z. Anorg. Allg. Chem. 2011, 637, 263–268. [Google Scholar] [CrossRef]
- Sergienko, V.S.; Aleksandrov, G.G.; Afonin, E.G. An unusual function of the anion of 1-hydroxyethane-1,1-diphosphonic acid (H4L): Crystal structure of [Ni(phen)3](H7L2)0.5(H5L2)0.5·2H2O. Cryst. Rep. 2000, 45, 432–438. [Google Scholar] [CrossRef]
- Yang, J.; Ma, J.-F.; Zheng, G.-L.; Li, L.; Li, F.-F.; Zhang, Y.-M.; Liu, J.-F. Divalent transition metal phosphonates with new structure containing hydrogen-bonded layers of phosphonate anions. J. Solid State Chem. 2003, 174, 116–124. [Google Scholar] [CrossRef]
- Latham, Κ.; Coyle, A.M.; Rix, C.J.; Fowless, A.; White, J.M. Effect of ring substituents on crystal packing and H-bonding in a series of halobis(phen)copper(II) arylphosphonic acid complexes. Polyhedron 2007, 26, 222–236. [Google Scholar] [CrossRef]
- Ma, K.-R.; Wei, C.-L.; Zhang, Y.; Kan, Y.-H.; Cong, M.-H.; Yang, X.-J. Structures and spectroscopy studies of two M(II)-phosphonate coordination polymers based on alkaline earth metals (M = Ba, Mg). J. Spectroscopy 2013, 2013. [Google Scholar] [CrossRef]
- Demadis, K.D.; Barouda, E.; Zhao, H.; Raptis, R.G. Structural architectures of charge-assisted, hydrogen-bonded, 2D layered amine···tetraphosphonate and zinc···tetraphosphonate ionic materials. Polyhedron 2009, 28, 3361–3367. [Google Scholar] [CrossRef]
- Murugavel, R.; Kuppuswamy, S.; Randoll, S. Cooperative binding of phosphate anion and a neutral nitrogen donor to alkaline-earth metal ions. Investigation of Group 2 metal-organophosphate interaction in the absence and presence of 1,10-phenanthroline. Inorg. Chem. 2008, 47, 6028–6039. [Google Scholar] [CrossRef]
- Lee, B.H.; Lynch, V.M.; Cao, G.; Mallouk, T.E. Structure of [Mg{HO3PCH(C6H5)2}2]·8H2O, a layered phosphonate salt. Acta Cryst. 1988, C44, 365–367. [Google Scholar]
- Guerri, A.; Taddei, M.; Bataille, T.; Moneti, S.; Boulon, M.-E.; Sangregorio, C.; Costantino, F.; Ienco, A. Same not the same: Thermally driven transformation of nickel phosphinate-bipyridine one-dimensional chains into three-dimensional coordination polymers. Cryst. Growth Des. 2018, 18, 2234–2242. [Google Scholar] [CrossRef]
- Schier, A.; Gamper, S.; Müller, G. Synthesis and crystal structure of magnesium bis[2-aminoethyl(hydrogen)phosphonate] octahydrate, Mg(2-AEPH)2·8H2O. Inorg. Chim. Acta 1990, 177, 179–183. [Google Scholar] [CrossRef]
- Smith, G.; Wermuth, U.D. The coordination complex structures and hydrogen bonding in the three-dimensional alkaline earth metal salts (Mg, Ca, Sr and Ba) of (4-aminophenyl)arsonic acid. Acta Cryst. 2017, C73, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Bekensir, A. Aromatic cation activation: Nucleophilic substitution of alcohols and carboxylic acids. Org. Lett. 2014, 16, 1720–1723. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.D.; Kotoris, C.C.; Dinaut, A.N.; Chen, M.-J. Synthesis of aryl(difluoromethylenephosphonates) via electrophilic fluorination of α-carbanions of benzylic phosphonates with N-fluorobenzenesulfonimide. Tetrahedron 1998, 54, 1691–1714. [Google Scholar] [CrossRef]
- Penicaud, V.; Maillet, C.; Janvier, P.; Pipelier, M.; Bujoli, B. New water-soluble diamine complexes as catalysts for the hydrogenation of ketones under hydrogen pressure. Eur. J. Org. Chem. 1999, 7, 1745–1748. [Google Scholar]
- Wydysh, E.A.; Medghalchi, S.M.; Vadlamudi, A.; Townsendd, C.A. Design and synthesis of small molecule glycerol 3-phosphate acyltransferase inhibitors. J. Med. Chem. 2009, 52, 3317–3327. [Google Scholar] [CrossRef] [PubMed]
- APEX3, V2018.7-2; Bruker AXS, Inc.: Madison, WI, USA, 2018.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 339–341. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- APEX2 v2014.9-0; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5653. [Google Scholar] [CrossRef]
- King, H.F.; Dupuis, M. Numerical integration using rys polynomials. J. Comput. Phys. 1976, 21, 144–165. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. Parts 1-4. J. Chem. Phys. 1955, 23, 1833–1840, 1841–1846, 2338–2342, and 2343–2346. [Google Scholar] [CrossRef]
- Lowdin, P.-O. On the orthogonality problem. Adv. Chem. Phys. 1970, 5, 185–199. [Google Scholar]
- Li, H.; Pomelli, C.; Jensen, J.H. Continuum solvation of large molecules described by QM/MM: A semi-iterative implementation of the PCM/EFP interface. J. Theor. Chem. Acc. 2003, 109, 71–84. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in electronic structure theory: GAMESS a decade later. In Theory and Applications of Computational Chemistry, the First Forty Years; Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherland, 2005; Chapter 41; pp. 1167–1189. [Google Scholar]
- Bode, B.M.; Gordon, M.S. MacMolPlt: a graphical user interface for GAMESS. J. Mol. Graphics Mod. 1998, 16, 133–138. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard III, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Baker, J. An algorithm for the location of transition states. J. Comput. Chem. 1986, 7, 385–395. [Google Scholar] [CrossRef]
- Helgaker, T. Transition-state optimizations by trust-region image minimization. Chem. Phys. Lett. 1991, 182, 503–510. [Google Scholar] [CrossRef]
- Culot, P.; Dive, G.; Nguyen, V.H.; Ghuysen, J.M. A quasi-Newton algorithm for first-order saddle-point location. Theoret. Chim. Acta 1992, 82, 189–205. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Demadis, K.D.; Papadaki, M.; Raptis, R.G.; Zhao, H. Corrugated, sheet-like architectures in layered alkaline earth metal R,S-hydroxyphosphonoacetate frameworks: Applications for anti-corrosion protection of metal surfaces. Chem. Mater. 2008, 20, 4835–4846. [Google Scholar] [CrossRef]
- Lodhia, S.; Turner, A.; Papadaki, M.; Demadis, K.D.; Hix, G.B. Polymorphism, composition and structural variability in topology in 1D, 2D and 3D copper phosphonocarboxylate materials. Cryst. Growth Des. 2009, 9, 1811–1822. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Rodriguez-Navarro, C.; Sebastian-Pardo, E. Sodium sulfate crystallization in the presence of phosphonates: Implications in ornamental stone conservation. Cryst. Growth Des. 2006, 6, 1575–1583. [Google Scholar] [CrossRef]
- Demadis, K.D.; Anagnostou, Z.; Panera, A.; Mezei, G.; Kirillova, M.V.; Kirillov, A.M. Three-Component 1D and 2D metal phosphonates: Structural variability, topological analysis and catalytic hydrocarboxylation of alkanes. RSC-Adv. 2017, 7, 17788–17799. [Google Scholar] [CrossRef]
- Demadis, K.D.; Armakola, E.; Papathanasiou, K.E.; Mezei, G.; Kirillov, A.M. Structural systematics and topological analysis of coordination polymers with divalent metals and a glycine-derived tripodal phosphonocarboxylate. Cryst. Growth Des. 2014, 14, 5234–5243. [Google Scholar] [CrossRef]
- Papathanasiou, K.E.; Demadis, K.D. Phosphonates in matrices. In Tailored Organic-Inorganic Materials; Brunet, E., Clearfield, A., Colon, J.L., Eds.; John Wiley & Sons Inc.: New York, NY, USA, 2015; Chapter 3; pp. 83–135. [Google Scholar]
- Demadis, K.D.; Theodorou, I.; Paspalaki, M. Controlled release of bis-phosphonate pharmaceuticals from cationic biodegradable polymeric matrices. Ind. Eng. Chem. Res. 2011, 50, 5873–5876. [Google Scholar] [CrossRef]
- Papathanasiou, K.E.; Turhanen, P.; Brückner, S.I.; Brunner, E.; Demadis, K.D. Smart, programmable and responsive injectable hydrogels for controlled release of cargo osteoporosis drugs. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Bazaga-García, M.; Papadaki, M.; Colodrero, R.M.P.; Olivera-Pastor, P.; Losilla, E.R.; Nieto-Ortega, B.; Aranda, M.A.G.; Choquesillo-Lazarte, D.; Cabeza, A.; Demadis, K.D. Tuning proton conductivity in alkali metal phosphonocarboxylates by cation size-induced and water-facilitated proton transfer pathways. Chem. Mater. 2015, 27, 424–435. [Google Scholar] [CrossRef]
- Salcedo, I.R.; Colodrero, R.M.P.; Bazaga-García, M.; Vasileiou, A.; Papadaki, M.; Olivera-Pastor, P.; Infantes-Molina, A.; Losilla, E.R.; Mezei, G.; Cabeza, A.; et al. From light to heavy alkali metal tetraphosphonates (M = Li, Na, K, Rb, Cs): Cation size-induced structural diversity and water-facilitated proton conductivity. CrystEngComm 2018, 20, 7648–7658. [Google Scholar] [CrossRef]
- Demadis, K.D.; Katarachia, S.D.; Zhao, H.; Raptis, R.G.; Baran, P. Alkaline earth metal organotriphosphonates: Inorganic-organic polymeric hybrids from dication-dianion association. Cryst. Growth Des. 2006, 6, 836–838. [Google Scholar] [CrossRef]
- Zorlu, Y.; Erbahar, D.; Çetinkaya, A.; Bulut, A.; Erkal, T.S.; Yazaydin, A.O.; Beckmann, J.; Yücesan, G. A cobalt arylphosphonate MOF – superior stability, sorption and magnetism. Chem. Commun. 2019, 55, 3053–3056. [Google Scholar] [CrossRef]
- Yücesan, G.; Zorlu, Y.; Stricker, M.; Beckmann, J. Metal-organic solids derived from arylphosphonic acids. Coord. Chem. Rev. 2018, 369, 105–122. [Google Scholar] [CrossRef]
- Silvestre, J.-P.; Dao, N.Q.; Leroux, Y. A survey of the behavior of the hydroxybisphosphonic function in crystallized acids, metallic salts, and some related compounds. Heteroatom Chem. 2001, 12, 73–89. [Google Scholar] [CrossRef]
- Beyer, O.; Homburg, T.; Albat, M.; Stock, N.; Lüning, U. Synthesis of phosphonosulfonic acid building blocks as linkers for coordination polymers. New J. Chem. 2017, 41, 8870–8876. [Google Scholar] [CrossRef]
- Schmidt, C.; Feyand, M.; Rothkirch, A.; Stock, N. High-throughput and in situ EDXRD investigation on the formation of two new metal aminoethylphosphonates–Ca(O3PC2H4NH2) and Ca(OH)(O3PC2H4NH3)·2H2O. J. Solid State Chem. 2012, 188, 44–49. [Google Scholar] [CrossRef]
- Gałȩzowska, J.; Czapor-Irzabek, H.; Janicki, R.; Chmielewska, E.; Janek, T. New aspects of coordination chemistry and biological activity of NTMP-related diphosphonates containing a heterocyclic ring. New J. Chem. 2017, 41, 10731–10741. [Google Scholar] [CrossRef]
- Shearan, S.; Stock, N.; Emmerling, F.; Demel, J.; Wright, P.A.; Demadis, K.D.; Vassaki, M.; Costantino, F.; Vivani, R.; Sallard, S.; et al. New directions in metal phosphonate and phosphinate chemistry. Crystals 2019, 9, 270. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mg-PABPA | Ca-PABPA | Sr-PABPA | Ni-EDPA | Ni-bpy-EDPA | Mg-TDTMP |
|---|---|---|---|---|---|---|
| Space group | Pc | C2/c | C2/c | P-1 | C2/c | P-1 |
| Chemical formula | C14H42MgN2O18P2 | C14H34CaN2O14P2 | C14H34N2O14P2Sr | C2H22NiO14P2 | C12H26N2NiO12P2 | C8H34MgN2O18P4 |
| Formula Mass (g/mol) | 612.74 | 556.45 | 603.99 | 390.84 | 511.00 | 594.56 |
| λ ( Å) | 0.71073 | 0.71073 | 1.54178 | 1.54178 | 0.71073 | 0.71073 |
| a (Å) | 7.2039(17) | 29.350(2) | 29.511(7) | 6.4856(4) | 16.953(1) | 5.8972(1) |
| b (Å) | 5.9781(13) | 6.2111(4) | 6.2928(13) | 6.5560(3) | 14.810(1) | 8.9705(1) |
| c (Å) | 31.547(7) | 12.9940(7) | 13.122(3) | 10.1363(5) | 10.5126(9) | 11.8486(1) |
| α (°) | 90 | 90 | 90 | 89.192(3) | 90.000 | 73.577(1) |
| β (°) | 91.521(9) | 107.220(3) | 106.947(7) | 73.902(3) | 127.430(3) | 76.201(1) |
| γ (°) | 90 | 90 | 90 | 62.131(3) | 90.000 | 75.534(1) |
| V (Å3) | 1358.1(5) | 2262.5(3) | 2331.0(9) | 362.66(3) | 2096.0(3) | 572.58(1) |
| Crystal size (mm) | 0.13 × 0.10 × 0.08 | 0.12 × 0.10 × 0.10 | 0.10 × 0.10 × 0.08 | 0.12 × 0.11 × 0.11 | 0.37 × 0.13 × 0.12 | 0.45 × 0.20 × 0.15 |
| Z | 2 | 4 | 4 | 1 | 4 | 1 |
| ρcalc (g·cm-3) | 1.498 | 1.634 | 1.721 | 1.790 | 1.619 | 1.724 |
| 2θ range (°) | 2.583–25.043 | 2.906–27.523 | 3.131–66.167 | 4.583–66.836 | 2.04–38.60 | 1.82–33.17 |
| Data/Restrains/ Parameters | 4709/2/339 | 2604/0/151 | 2018/0/155 | 1259/0/99 | 5872/5/150 | 4349/8/175 |
| Nº reflections | 21287 | 23620 | 11945 | 4686 | 46140 | 49091 |
| Independent reflections [I > 2σ(I)] | 4709 | 2604 | 2018 | 1259 | 5090 | 4043 |
| GoF | 1.096 | 1.019 | 1.137 | 1.056 | 1.042 | 1.443 |
| R Factor [I > 2σ(I)] | aR1 = 0.0762, awR2 = 0.1948 | aR1 = 0.0397, awR2 = 0.0811 | aR1 = 0.0460, awR2 = 0.1304 | aR1 = 0.0464, awR2 = 0.1189 | aR1 = 0.0262 awR2 = 0.0643 | aR1 = 0.0277 awR2 = 0.0905 |
| R Factor (all data) | aR1 = 0.1015, awR2 = 0.2059 | aR1 = 0.0695, awR2 = 0.0864 | aR1 = 0.0483, awR2 = 0.1330 | aR1 = 0.0480, awR2 = 0.1203 | aR1 = 0.0333 awR2 = 0.0679 | aR1 = 0.0299 awR2 = 0.0923 |
| CCDC Code | 1914866 | 1914867 | 1914868 | 1914870 | 1914871 | 1914872 |
| HBPA1− | BPA2− | ||||
|---|---|---|---|---|---|
| Atom # | δq Mulliken | δq Lowdin | Atom # | δq Mulliken | δq Lowdin |
| 16 | −0.932911 | −0.884627 | 16 | −1.062429 | −0.999545 |
| 17 | −0.969105 | −0.891882 | 17 | −1.064163 | −1.005116 |
| 18 | −0.895888 | −0.796636 | 18 | −1.072408 | −0.986760 |
| mean | −0.932635 | −0.857715 | mean | −1.066333 | −0.997140 |
| (HPABPA1−)2 | HPABPA1− | ||||
|---|---|---|---|---|---|
| Atom # | δq Mulliken | δq Lowdin | Atom # | δq Mulliken | δq Lowdin |
| 2 | −1.011181 | −0.817501 | 15 | −1.024967 | −0.976540 |
| 3 | −0.946950 | −0.905140 | 16 | −1.062499 | −0.994478 |
| 4 | −0.982415 | −0.894151 | 17 | −1.050593 | −0.974468 |
| 23 | −0.975002 | −0.898125 | |||
| 24 | −0.936067 | −0.911068 | |||
| 25 | −0.974519 | −0.821586 | |||
| mean | −0.971022 | −0.874595 | mean | −1.046020 | −0.981829 |
| H2EDPA2− | ||
|---|---|---|
| Atom # | δq Mulliken | δq Lowdin |
| 9 | −0.972095 | −0.893946 |
| 10 | −0.961244 | −0.900770 |
| 11 | −0.936159 | −0.814461 |
| 13 | −0.974152 | −0.889730 |
| 14 | −0.959120 | −0.811834 |
| 16 | −0.956155 | −0.905960 |
| mean | −0.959821 | −0.869450 |
| HEPA1− | EPA2− | ||||
|---|---|---|---|---|---|
| Atom # | δq Mulliken | δq Lowdin | Atom # | δq Mulliken | δq Lowdin |
| 9 | −0.965273 | −0.893808 | 9 | −1.101121 | −1.004788 |
| 10 | −0.964247 | −0.819322 | 10 | −1.103426 | −1.014740 |
| 12 | −0.991020 | −0.907261 | 11 | −1.116413 | −1.012214 |
| mean | −0.973513 | −0.873464 | mean | −1.106987 | −1.010581 |
| Molecules Pair | % Charge Reduction | |
|---|---|---|
| Mulliken | Lowdin | |
| HBPA1− - HPABPA1− | −12% | −15% |
| BPA2− - HPABPA1− | +2% | +2% |
| HBPA1− - (HPABPA1−)2 | −4% | −2% |
| BPA2− - (HPABPA1−)2 | +9% | +12% |
| HPABPA1− - (HPABPA1−)2 | +7% | +11% |
| HEPA1− - H2EDPA2− | +1% | +1% |
| EPA2− - H2EDPA2− | +13% | +14% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xanthopoulos, K.; Anagnostou, Z.; Chalkiadakis, S.; Choquesillo-Lazarte, D.; Mezei, G.; Zaręba, J.K.; Zoń, J.; Demadis, K.D. Platonic Relationships in Metal Phosphonate Chemistry: Ionic Metal Phosphonates. Crystals 2019, 9, 301. https://doi.org/10.3390/cryst9060301
Xanthopoulos K, Anagnostou Z, Chalkiadakis S, Choquesillo-Lazarte D, Mezei G, Zaręba JK, Zoń J, Demadis KD. Platonic Relationships in Metal Phosphonate Chemistry: Ionic Metal Phosphonates. Crystals. 2019; 9(6):301. https://doi.org/10.3390/cryst9060301
Chicago/Turabian StyleXanthopoulos, Konstantinos, Zafeiria Anagnostou, Sophocles Chalkiadakis, Duane Choquesillo-Lazarte, Gellert Mezei, Jan K. Zaręba, Jerzy Zoń, and Konstantinos D. Demadis. 2019. "Platonic Relationships in Metal Phosphonate Chemistry: Ionic Metal Phosphonates" Crystals 9, no. 6: 301. https://doi.org/10.3390/cryst9060301
APA StyleXanthopoulos, K., Anagnostou, Z., Chalkiadakis, S., Choquesillo-Lazarte, D., Mezei, G., Zaręba, J. K., Zoń, J., & Demadis, K. D. (2019). Platonic Relationships in Metal Phosphonate Chemistry: Ionic Metal Phosphonates. Crystals, 9(6), 301. https://doi.org/10.3390/cryst9060301