Crystallization Kinetics of Polyamide 12 during Selective Laser Sintering

Abstract
:1. Introduction
- First, the building platform is lowered to a layer thickness of about 100 µm and the pre-heated powder is applied into the building chamber with a roller or blade.
- Second, the semi-crystalline thermoplastic powder is heated to a temperature just below the melting point. When the building chamber temperature is reached, depending on the given layer geometry, it connects to the individual layer below, and a CO2 laser, which is guided over the scanner mirror, selectively melts the preheated powder particles. The laser should only introduce the necessary amount of heat to melt a layer, to ensure the connection to the lower layer, and to minimize warpage during cooling.
- Last, in the subsequent consolidation phase, the melt is cooled again to the building chamber temperature; crystallization of the powder melt should not occur in this phase. This results in the formation of a two-phase mixing region in which the surrounding powder has approximately the same temperature as the molten powder. The surrounding powder, which is not melted, serves a support function and also serves as an insulating layer and is intended to ensure uniform cooling and thus homogeneous crystallization [2]. The described steps are repeated until the component is finished.
1.1. Isothermal Crystallization Kinetic Model
1.2. Non-Isothermal Crystallization Kinetic Model
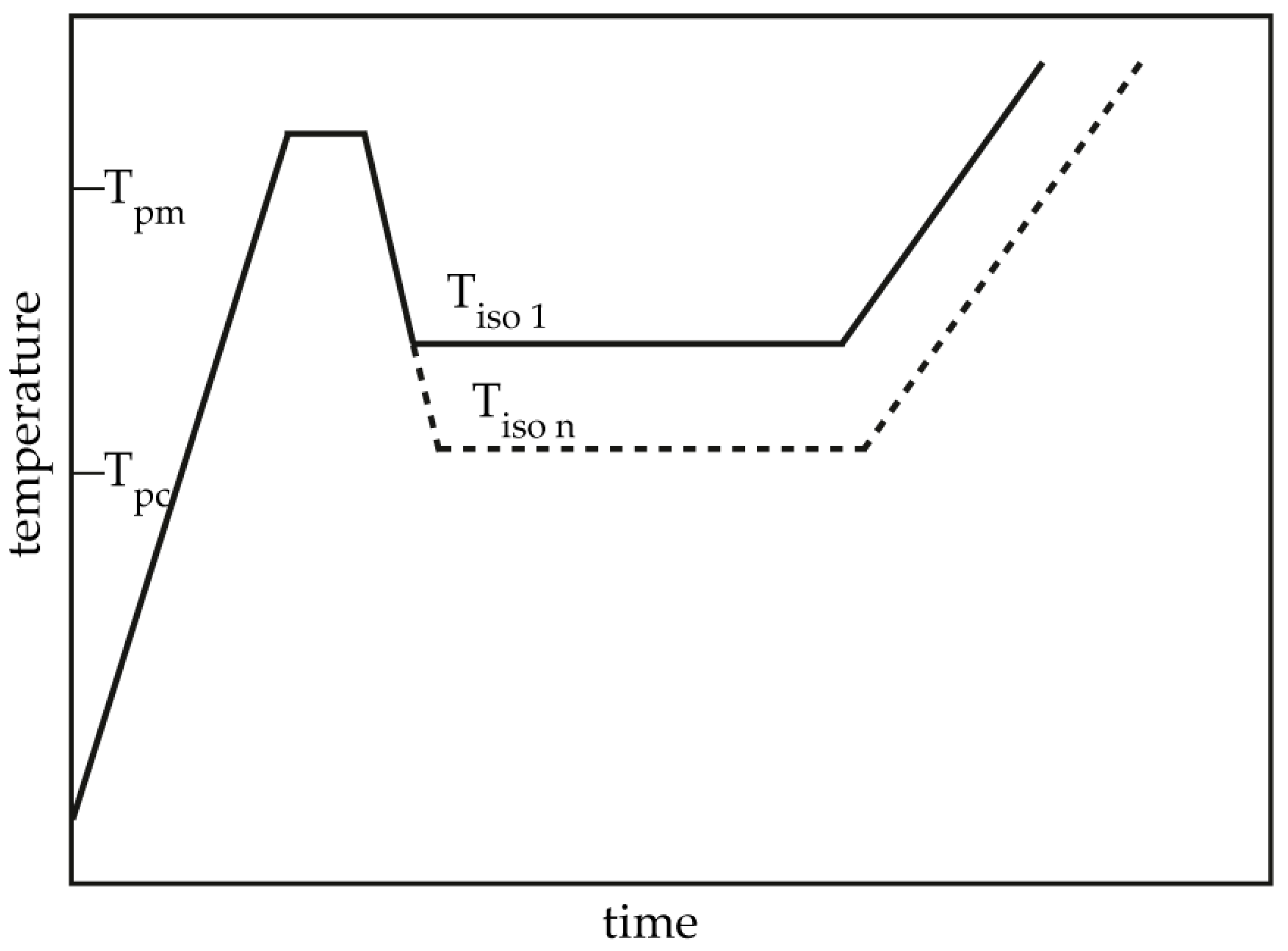
2. Materials and Methods
3. Results and Discussion
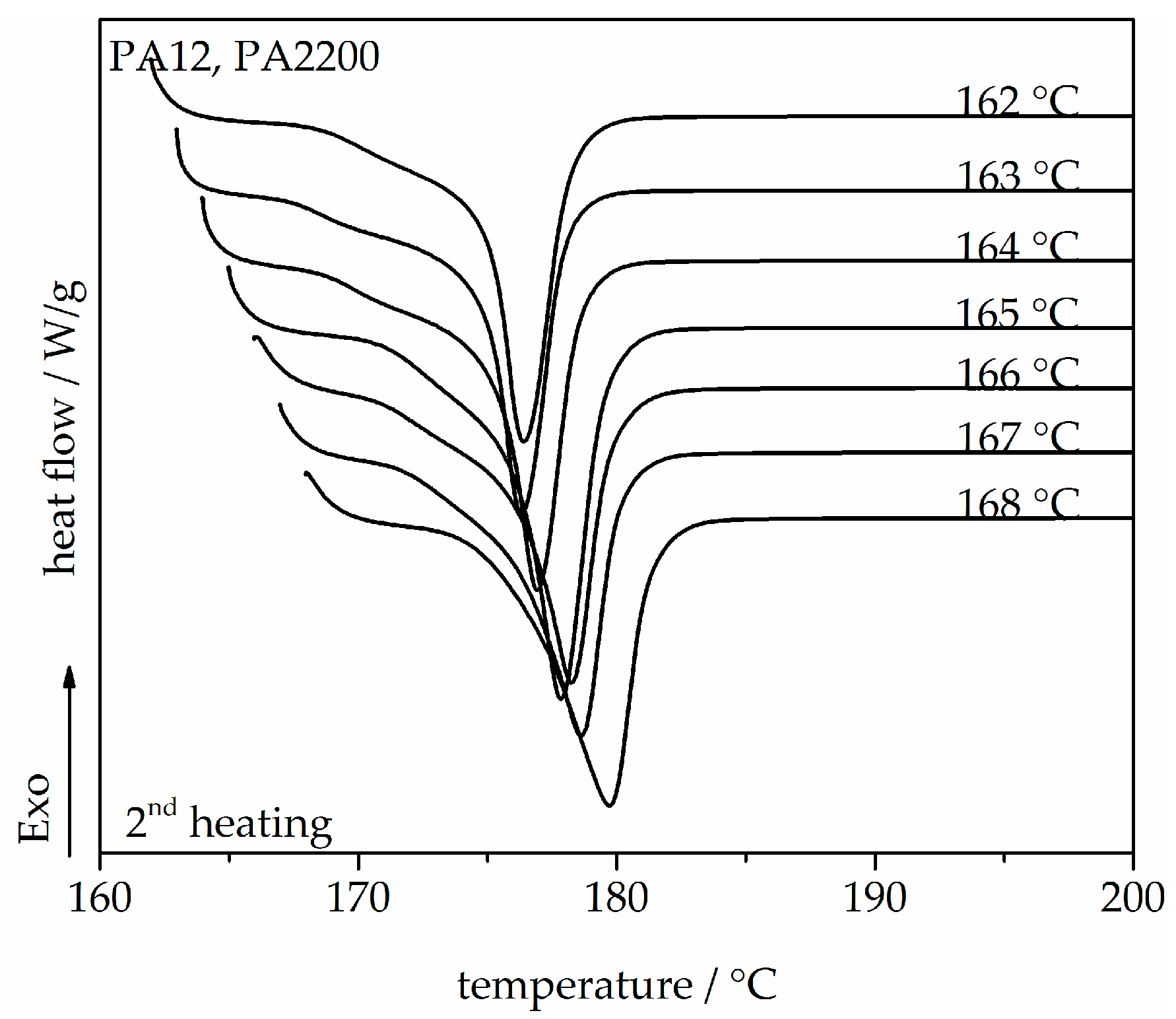
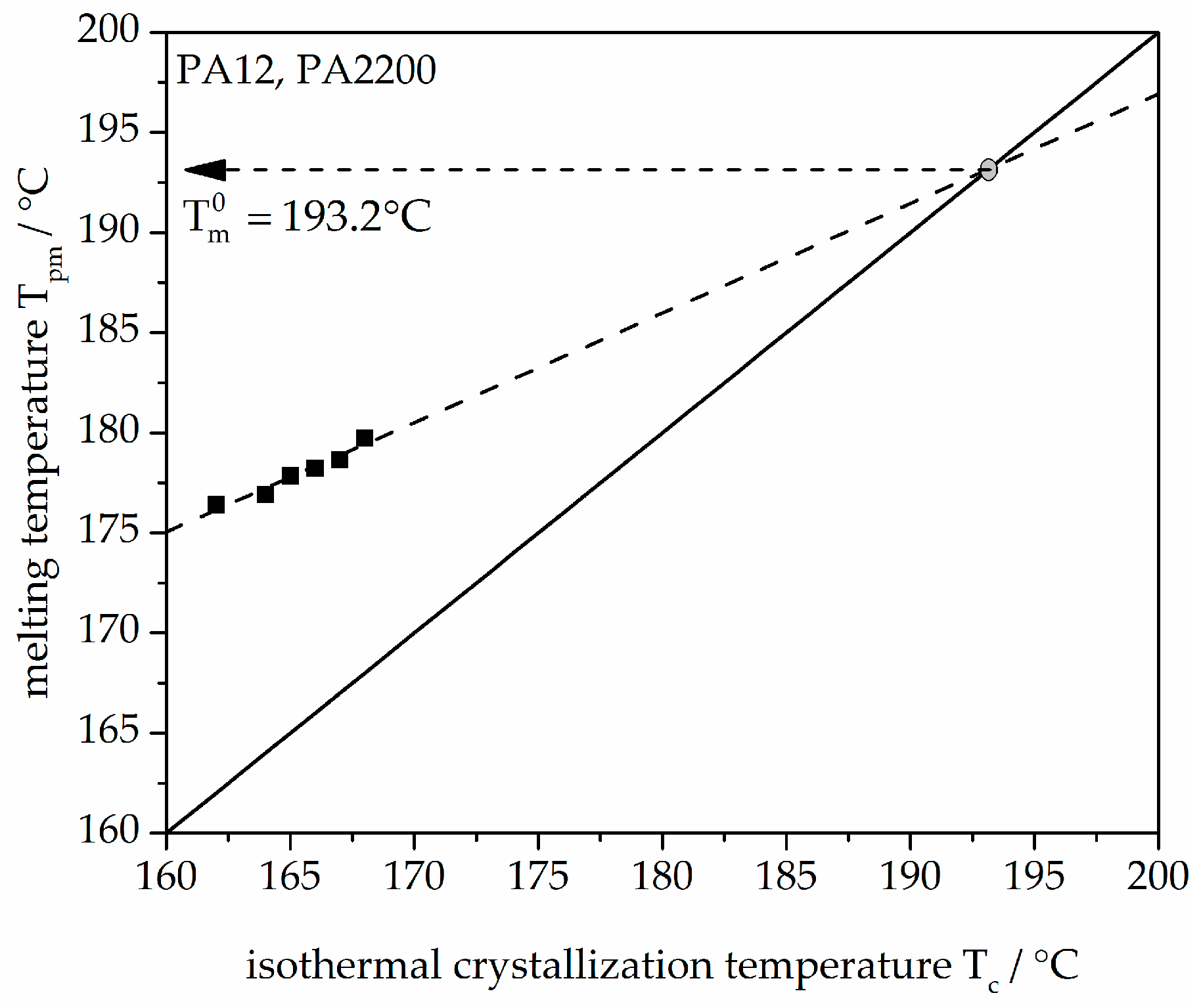
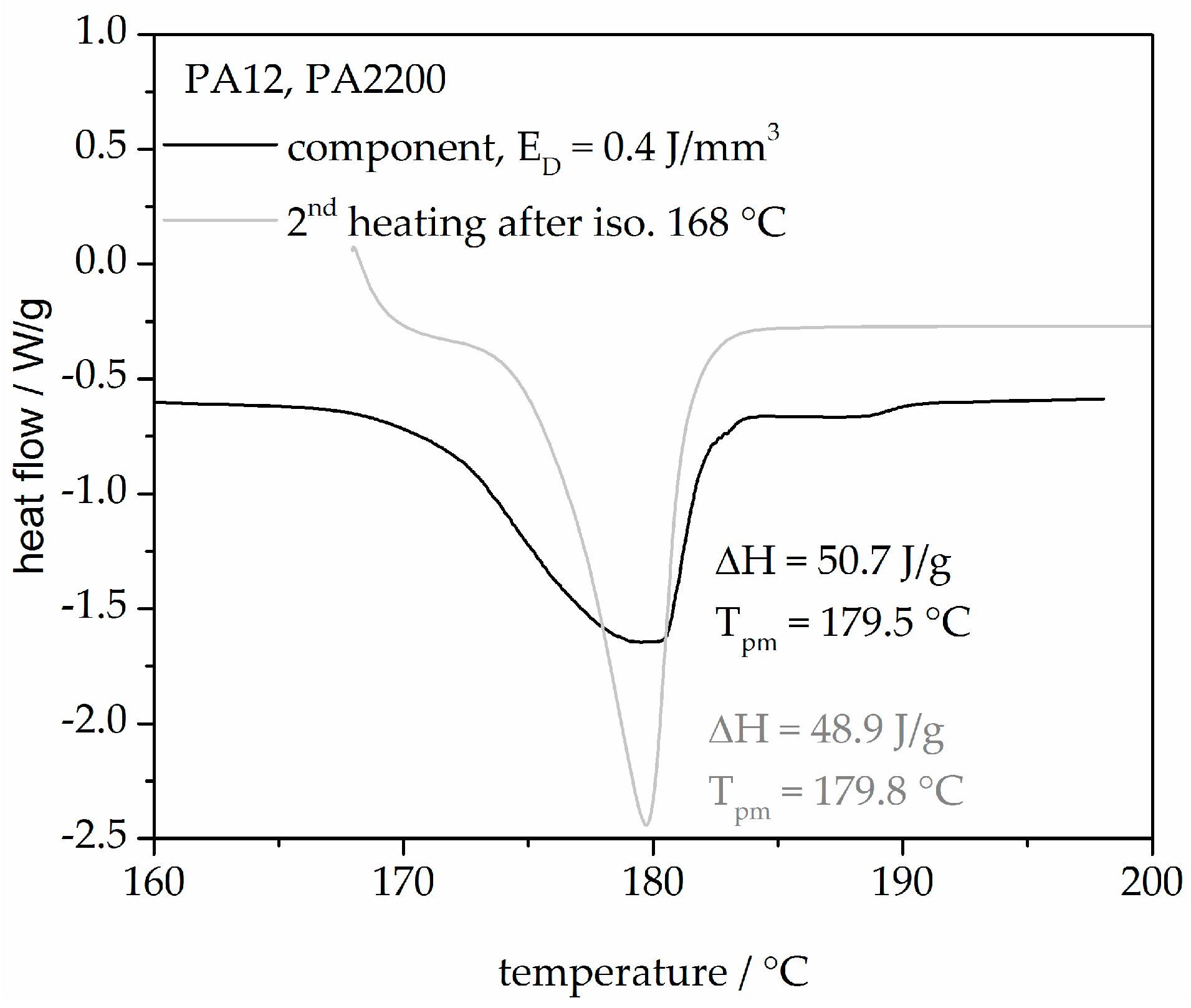
3.1. Melting Behavior and Equilibrium Melting Temperature
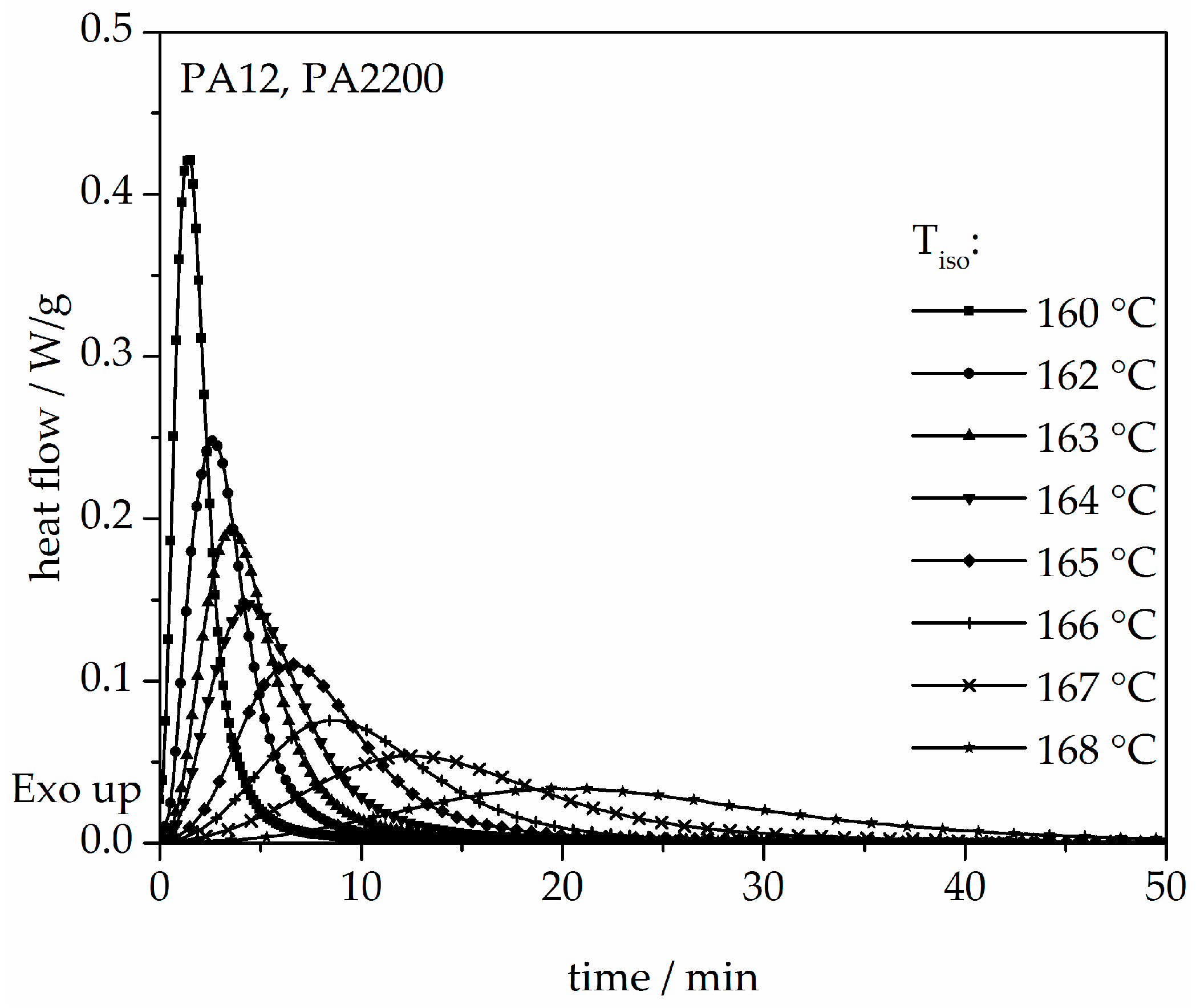
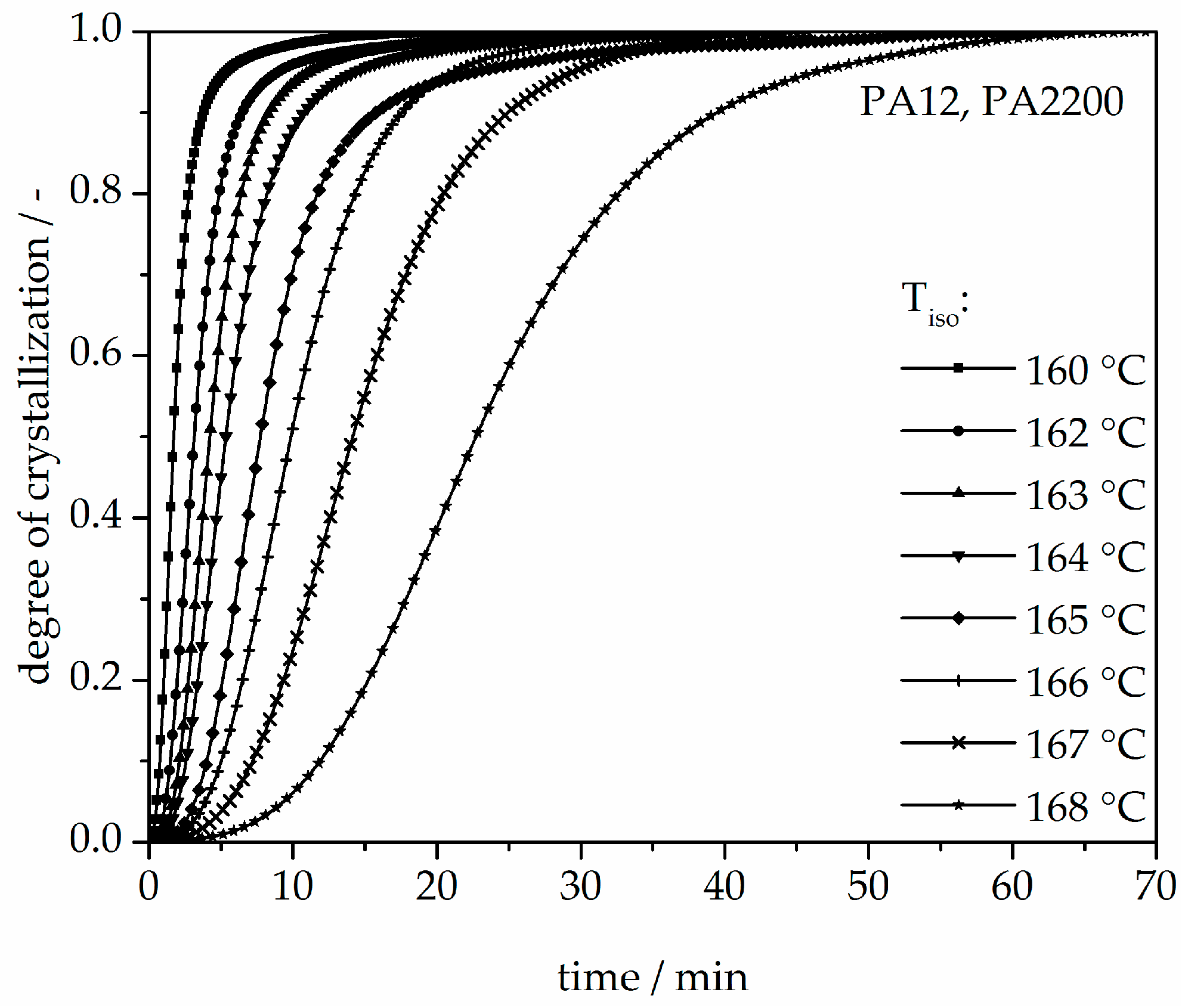
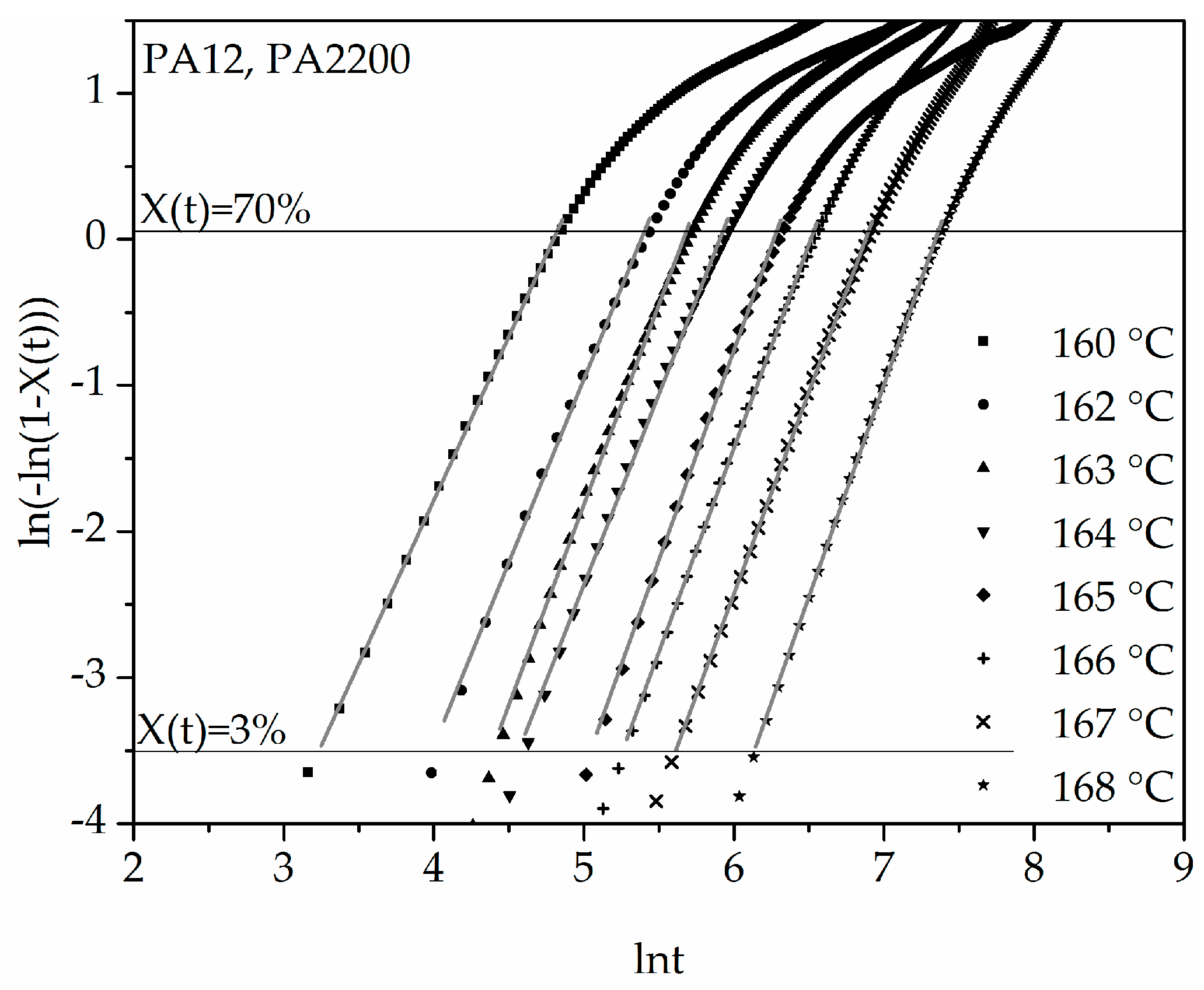
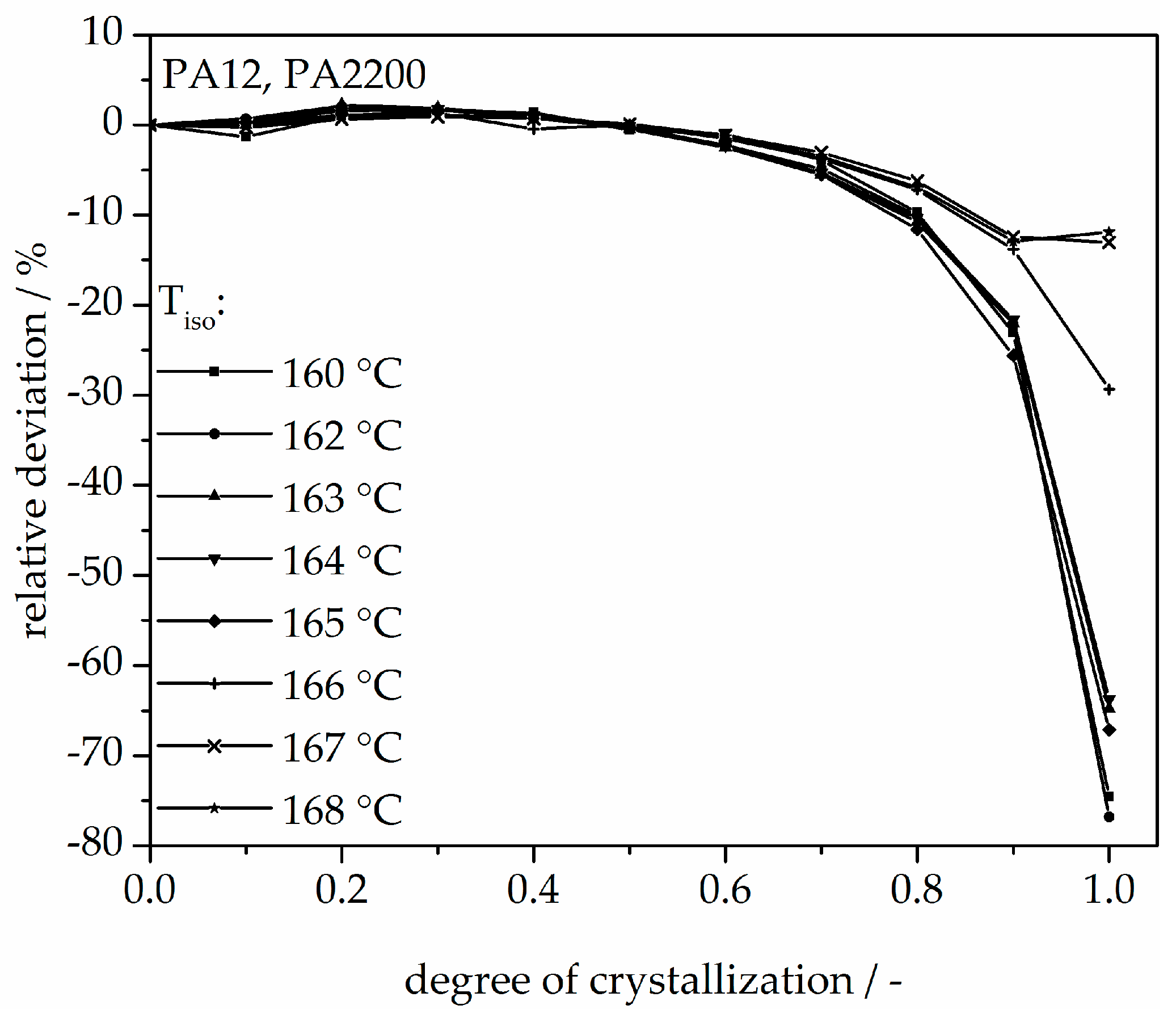
3.2. Isothermal Crystallization Kinetics Analysis
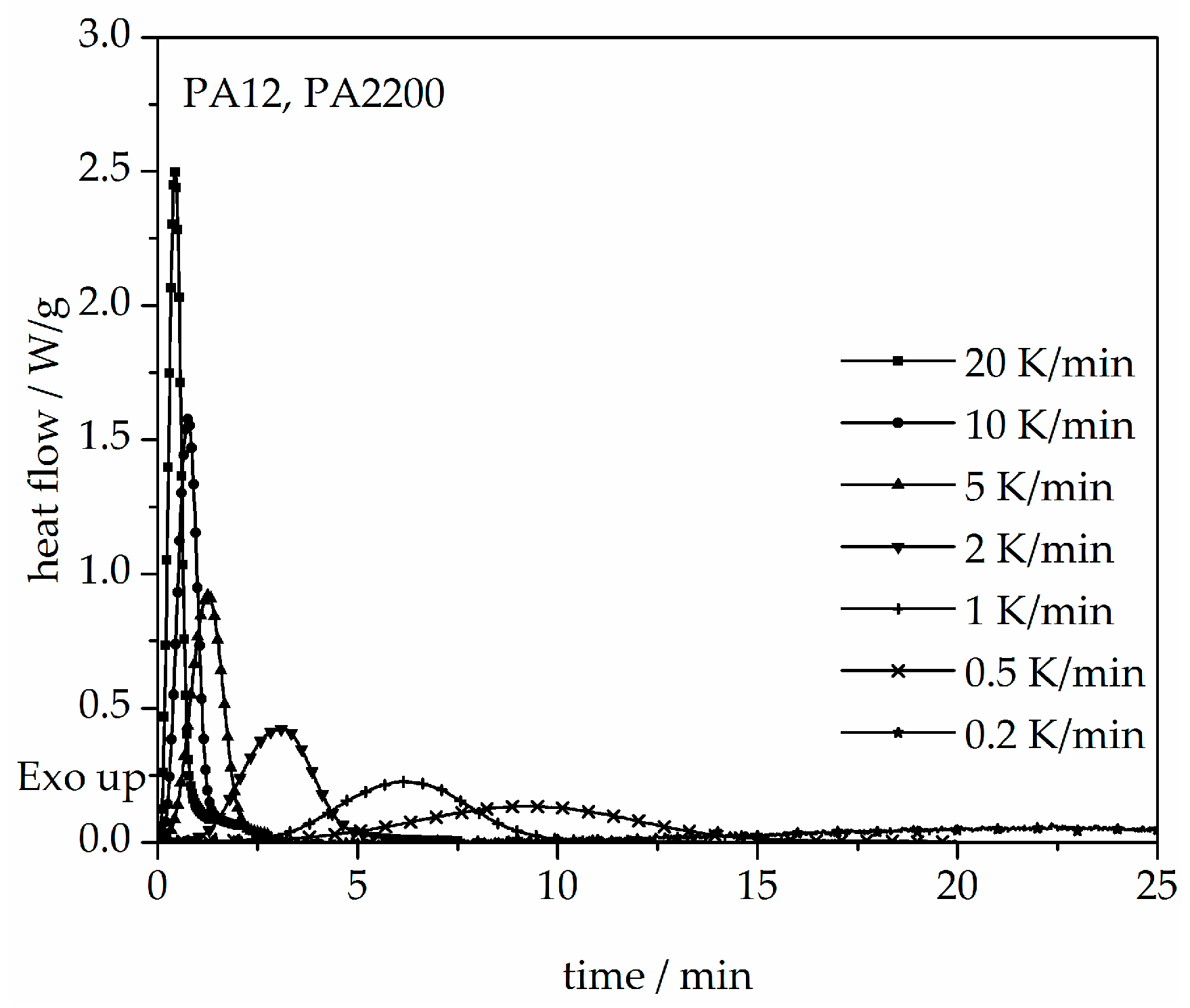
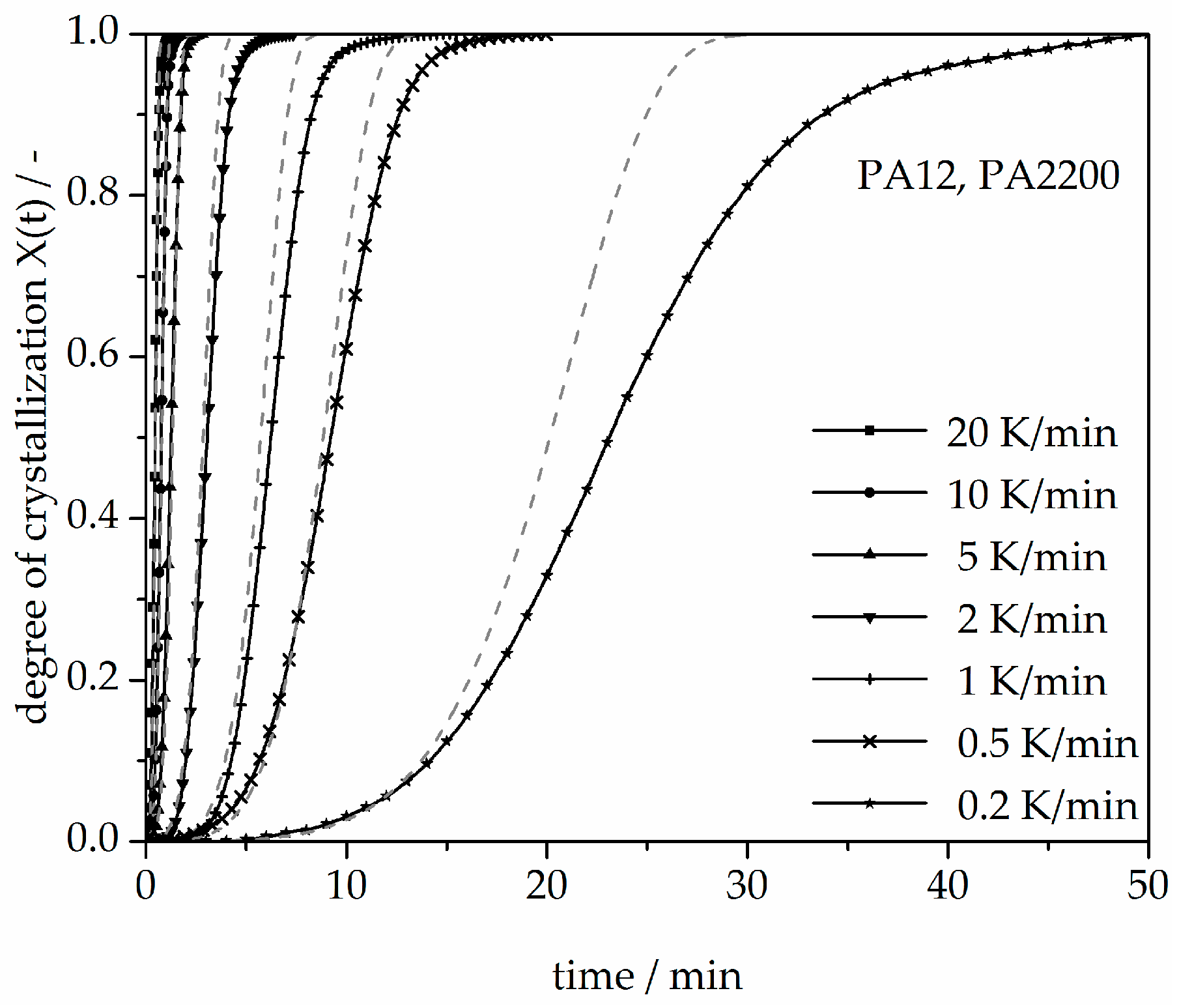
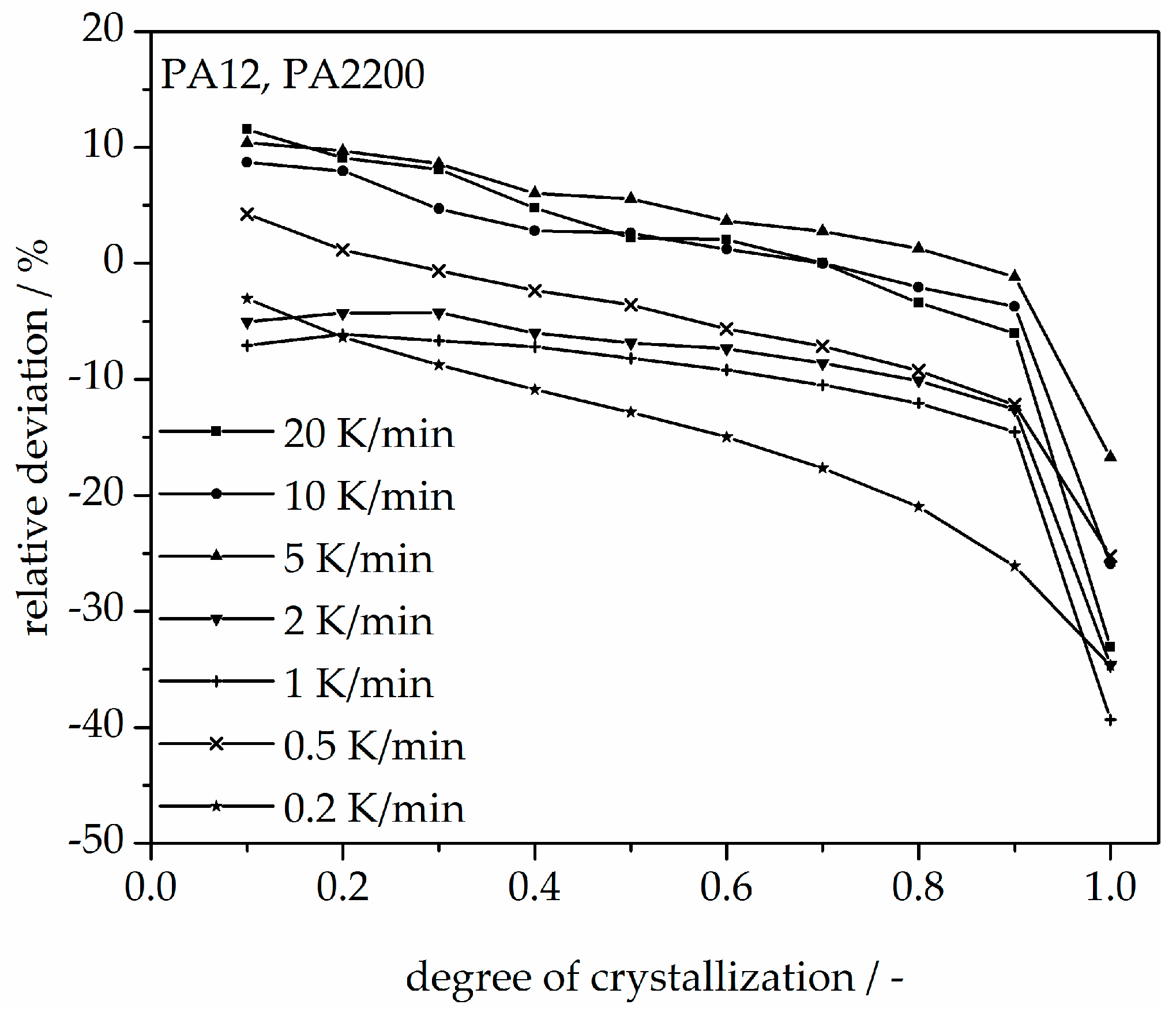
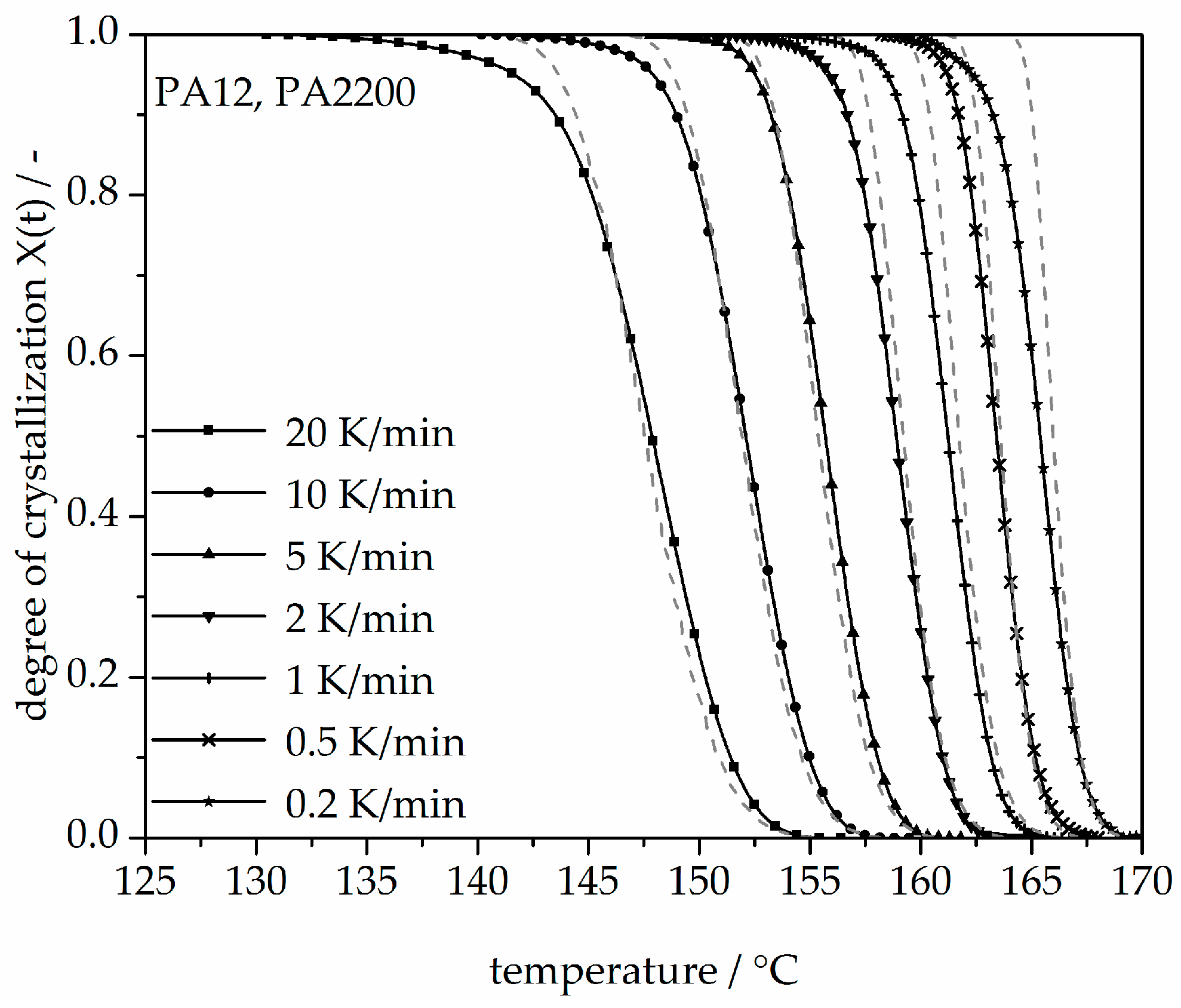
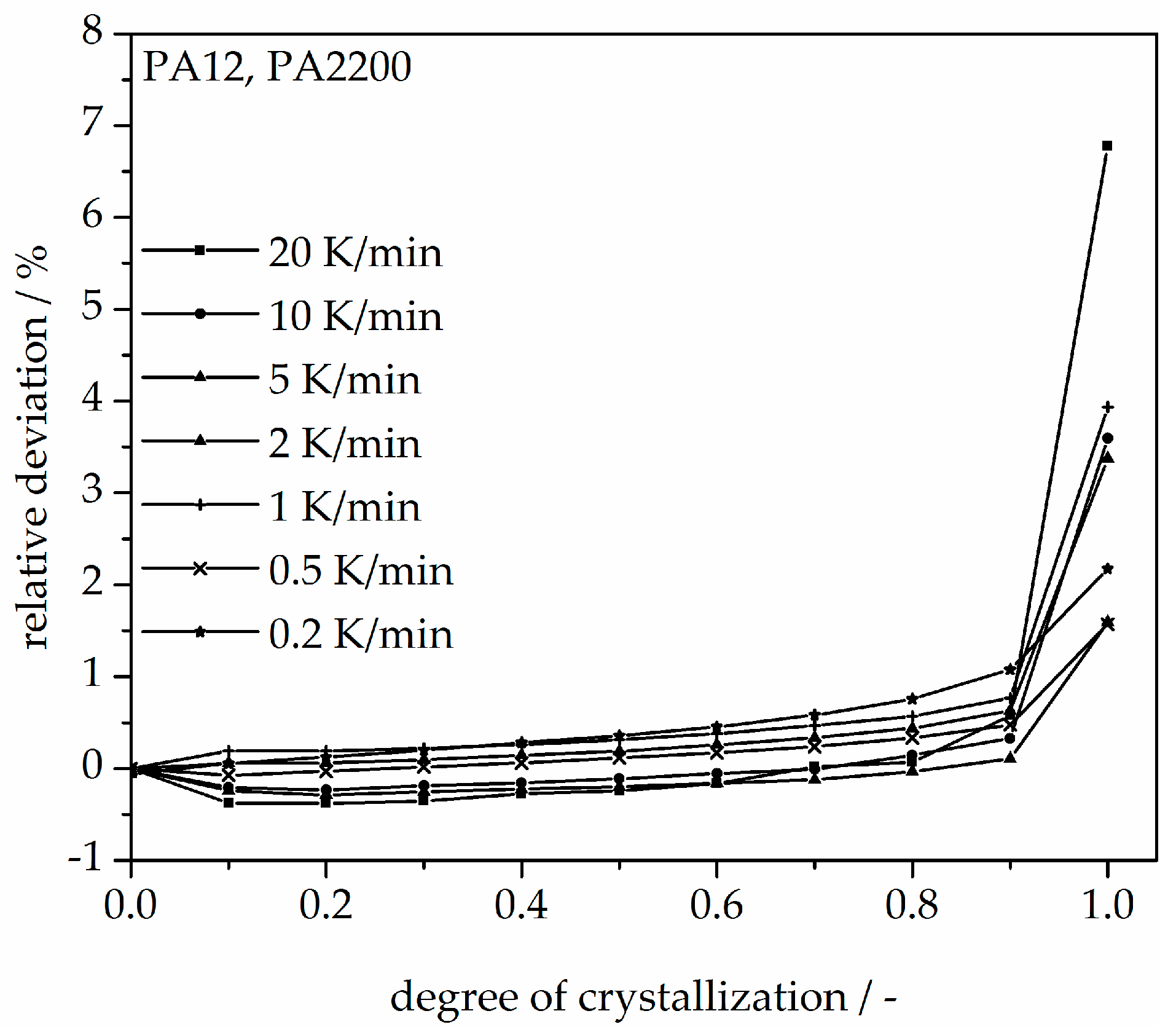
3.3. Non-Isothermal Crystallization Kinetic Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gibson, I.; Rosen, D.; Stucker, B. Additive Manufacturing Technologies: 3D Printing, Rapid Prototyping, and Direct Digital Manufacturing, 2nd ed.; Springer: New York, NY, USA, 2014; ISBN 9781493921133. [Google Scholar]
- Amado Becker, A.F. Characterization and Prediction of SLS Processability of Polymer Powders with Respect to Powder Flow and Part Warpage. Ph.D. Thesis, ETH Zurich, Zurich, Switzerland, 2016. [Google Scholar]
- Kruth, J.P.; Levy, G.; Schindel, R.; Craeghs, T.; Yasa, E. Consolidation of polymer powders by selective laser sintering. In Proceedings of the 3rd International Conference on Polymers and Moulds Innovations, Ghent, Belgium, 17–19 September 2008. [Google Scholar]
- Gebhardt, A. Understanding Additive Manufacturing: Rapid Prototyping-Rapid Tooling-Rapid Manufacturing, 1st ed.; Cark Hanser Verlag: Munich, Germany, 2012; ISBN 978-3-446-42552-1. [Google Scholar]
- Zarringhalam, H.; Hopkinson, N.; Kamperman, N.F.; de Vlieger, J.J. Effects of processing on microstructure and properties of SLS Nylon 12. Mater. Sci. Eng. A 2006, 435–436, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Wudy, K.; Drummer, D.; Kühnlein, F.; Drexler, M. Influence of degradation behavior of polyamide 12 powders in laser sintering process on produced parts. AIP Conf. Proc. 2014, 1593, 691. [Google Scholar] [CrossRef]
- Drummer, D.; Drexler, M.; Wudy, K. Impact of heating rate during exposure of laser molten parts on the processing window of PA12 powder. Phys. Procedia 2014, 56, 184–192. [Google Scholar] [CrossRef]
- Shen, J.; Steinberger, J.; Goepfert, R.; Gerner, F.; Daiber, K.; Manetsberger, S.F. Inhomogeneous shrinkage of polymer materials in selective laser sintering. In Proceedings of the Solid Freeform Fabrication Symposium, Austin, TX, USA, 9–11 August 2000. [Google Scholar]
- Plummer, C.J.G.; Zanetto, J.E.; Bourban, P.E.; Manson, J.A.E. The crystallization kinetics of polyamide-12. Colloid Polym. Sci. 2001, 279, 312–322. [Google Scholar] [CrossRef]
- Amado, A.; Wegener, K.; Schmid, M.; Levy, G. Characterization and modeling of non-isothermal crystallization of Polyamide 12 and co-Polypropylene during the SLS process. In Proceedings of the 5th International Polymers & Moulds Innovations Conference, Ghent, Belgium, 12–14 September 2012; pp. 207–216. [Google Scholar]
- Neugebauer, F.; Ploshikhin, V.; Ambrosy, J.; Witt, G. Isothermal and non-isothermal crystallization kinetics of polyamide 12 used in laser sintering. J. Therm. Anal. Calorim. 2016, 124, 925–933. [Google Scholar] [CrossRef]
- Liu, M.; Zhao, Q.; Wang, Y.; Zhang, C.; Mo, Z.; Cao, S. Melting behaviors, isothermal and non-isothermal crystallization kinetics of nylon 1212. Polymer 2003, 44, 2537–2545. [Google Scholar] [CrossRef]
- Piorkowska, E.; Rutledge, G.C. Handbook of Polymer Crystallization, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; ISBN 9780470380239. [Google Scholar]
- Avrami, M. Kinetics of phase change. I General theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar]
- Avrami, M. Kinetics of phase change. II Transformation-time relations for random distribution of nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Granulation, phase change, and microstructure kinetics of phase change. III. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Mandelkern, L. Crystallization of Polymers: Volume 2, Kinetics and Mechanisms, 2nd ed.; Cambridge University Press: Cambridge, UK, 2004; ISBN 0521816823. [Google Scholar]
- Ozawa, T. Kinetics of non-isothermal crystallization. Polymer 1971, 12, 150–158. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, C.; Li, H.; Li, Q.; Chen, J. Nonisothermal cold crystallization kinetics of poly (ethylene terephthalate)/clay nanocomposite. J. Appl. Polym. Sci. 2004, 91, 308–314. [Google Scholar] [CrossRef]
- Jeziorny, A. Parameters characterizing the kinetics of the non-isothermal crystallization of poly (ethylene terephthalate) determined by DSC. Polymer 1978, 19, 1142–1144. [Google Scholar] [CrossRef]
- Nakamura, K.; Katayama, K.; Amano, T. Some aspects of nonisothermal crystallization of polymers. II. Consideration of the isokinetic condition. J. Appl. Polym. Sci. 1973, 17, 1031–1041. [Google Scholar]
- Nakamura, K.; Watanabe, T.; Amano, T.; Katayama, K. Some aspects of nonisothermal crystallization of polymers. III. Crystallization during melt spinning. J. Appl.Polym. Sci. 1974, 18, 615–623. [Google Scholar]
- Nakamura, K.; Watanabe, T.; Katayama, K.; Amano, T. Some aspects of nonisothermal crystallization of polymers. I. Relationship between crystallization temperature, crystallinity, and cooling conditions. J. Appl. Polym. 1972, 16, 1077–1091. [Google Scholar] [CrossRef]
- Patel, R.M.; Spruiell, J.E. Crystallization kinetics during polymer processing—Analysis of available approaches for process modeling. Polym. Eng. Sci. 1991, 31, 730–738. [Google Scholar] [CrossRef]
- Ziabicki, A. Fundamentals of Fibre Formation: The Science of Fibre Spinning and Drawing; Wiley: Hoboken, NJ, USA, 1976; ISBN 0471982202. [Google Scholar]
- Zhao, M.; Drummer, D.; Wudy, K.; Drexler, M. Sintering Study of Polyamide 12 Particles for Selective Laser Melting. Int. J. Recent Contrib. Eng. Sci. IT (iJES) 2015, 3, 28–33. [Google Scholar] [CrossRef]
- Baldenegro-Perez, L.A.; Navarro-Rodriguez, D.; Medellin-Rodriguez, F.J.; Hsiao, B.; Avila-Orta, C.A.; Sics, I. Molecular Weight and Crystallization Temperature Effects on Poly(ethylene terephthalate) (PET) Homopolymers, an Isothermal Crystallization Analysis. Polymers 2014, 6, 583–600. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Weeks, J.J. Melting process and the equilibrium melting temperature of polychlorotrifluoroethylene. J. Res. Natl. Bur. Stand. A 1962, 66, 13–28. [Google Scholar] [CrossRef]
- Mark, J.E. Physical Properties of Polymers Handbook; Springer: Berlin, Germany, 2007; Volume 1076, ISBN 978-0-387-69002-5. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
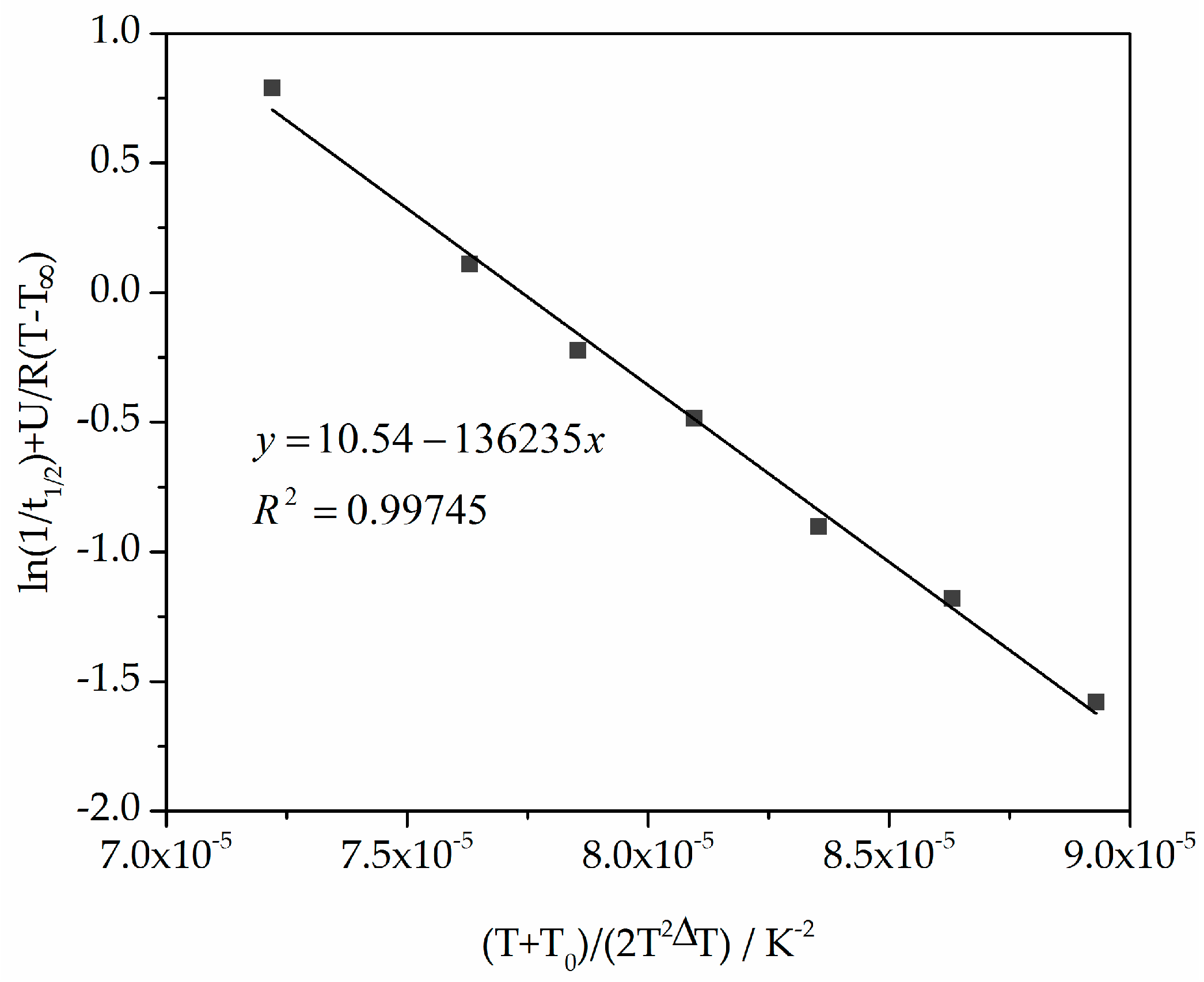{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tc °C | n | n [11] | k | k [11] | tmax | t1/2 Measurement | t1/2 Model |
|---|---|---|---|---|---|---|---|
| - | - | S−1 | s | s | s | ||
| 160 | 2.24 | 2.71 | 2.15 × 10−5 | 2.65 × 10−7 | 88.6 | 103.2 | 102.6 |
| 162 | 2.51 | 2.94 | 1.37 × 10−6 | 1.34 × 10−8 | 158.4 | 188.4 | 187.8 |
| 163 | 2.74 | 1.81 × 10−7 | 212.4 | 253.3 | 252.0 | ||
| 164 | 2.61 | 3.05 | 2.01 × 10−7 | 1.08 × 10−9 | 260.4 | 319.8 | 319.2 |
| 165 | 2.87 | 1.59 × 10−8 | 396.6 | 463.8 | 462.6 | ||
| 166 | 2.79 | 2.69 | 1.28 × 10−8 | 2.62 × 10−9 | 510.0 | 591.0 | 591.0 |
| 167 | 2.77 | 5.44 × 10−9 | 738.6 | 849.0 | 849.6 | ||
| 168 | 2.90 | 2.94 | 5.69 × 10−10 | 6.34 × 10−11 | 1179.0 | 1362.6 | 1362.6 |
| Φ K/min | Tic | tmax | t1/2 Measurement | t1/2 Model |
|---|---|---|---|---|
| °C | s | s | s | |
| 20 | 156.4 | 25.8 | 27.6 | 28.2 |
| 10 | 169.5 | 45.0 | 46.2 | 47.4 |
| 5 | 161.8 | 75.0 | 75.6 | 79.8 |
| 2 | 164.9 | 186.6 | 183.0 | 170.4 |
| 1 | 167.4 | 367.8 | 373.2 | 342.6 |
| 0.5 | 168.0 | 558.6 | 553.2 | 533.4 |
| 0.2 | 170.0 | 1341.0 | 1387.2 | 1208.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Wudy, K.; Drummer, D. Crystallization Kinetics of Polyamide 12 during Selective Laser Sintering. Polymers 2018, 10, 168. https://doi.org/10.3390/polym10020168
Zhao M, Wudy K, Drummer D. Crystallization Kinetics of Polyamide 12 during Selective Laser Sintering. Polymers. 2018; 10(2):168. https://doi.org/10.3390/polym10020168
Chicago/Turabian StyleZhao, Meng, Katrin Wudy, and Dietmar Drummer. 2018. "Crystallization Kinetics of Polyamide 12 during Selective Laser Sintering" Polymers 10, no. 2: 168. https://doi.org/10.3390/polym10020168
APA StyleZhao, M., Wudy, K., & Drummer, D. (2018). Crystallization Kinetics of Polyamide 12 during Selective Laser Sintering. Polymers, 10(2), 168. https://doi.org/10.3390/polym10020168