Kinetics of Ordering and Deformation in Photosensitive Azobenzene LC Networks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model and Main Equations
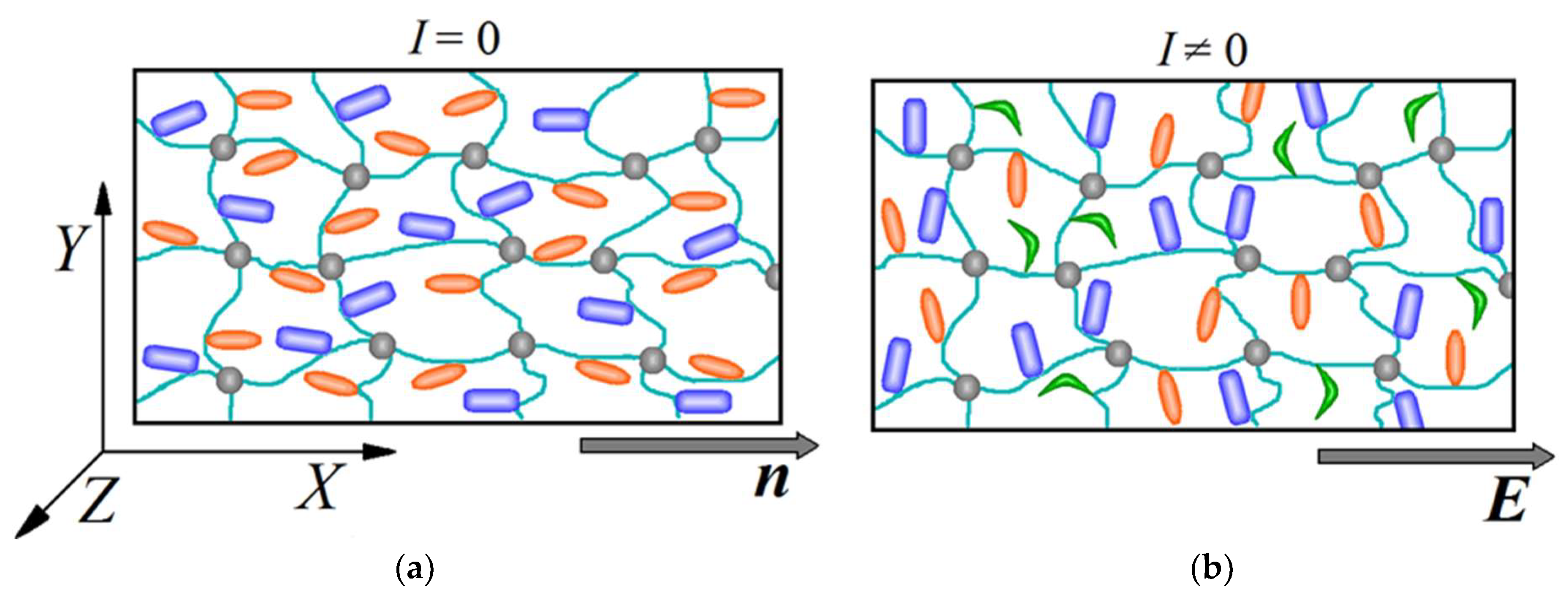
2.1. Light-Induced Deformation of an Azobenzene-Containing LC Network
2.2. Kinetic Equations of Photoisomerization
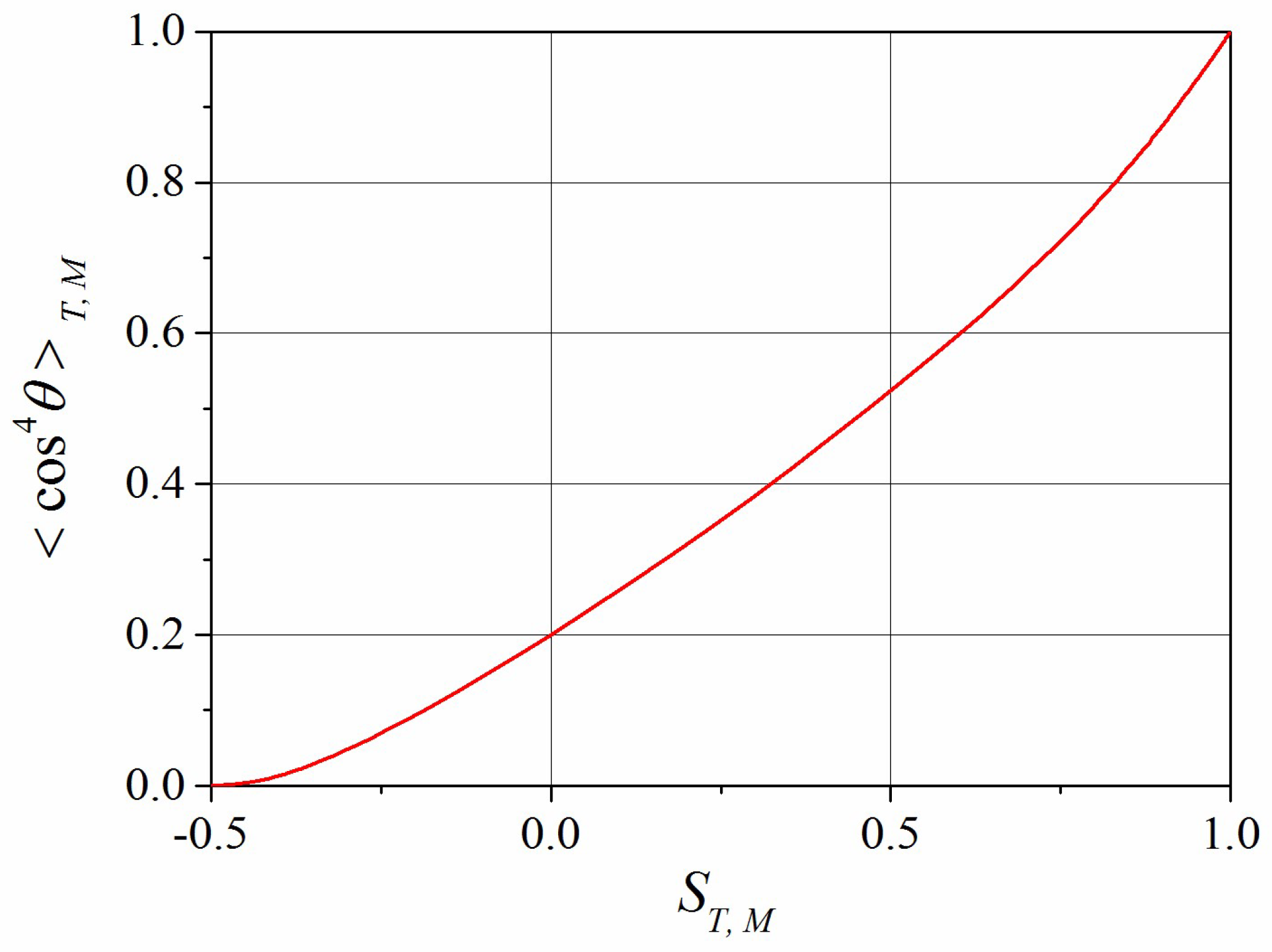
2.3. Closure Approximation
3. Results
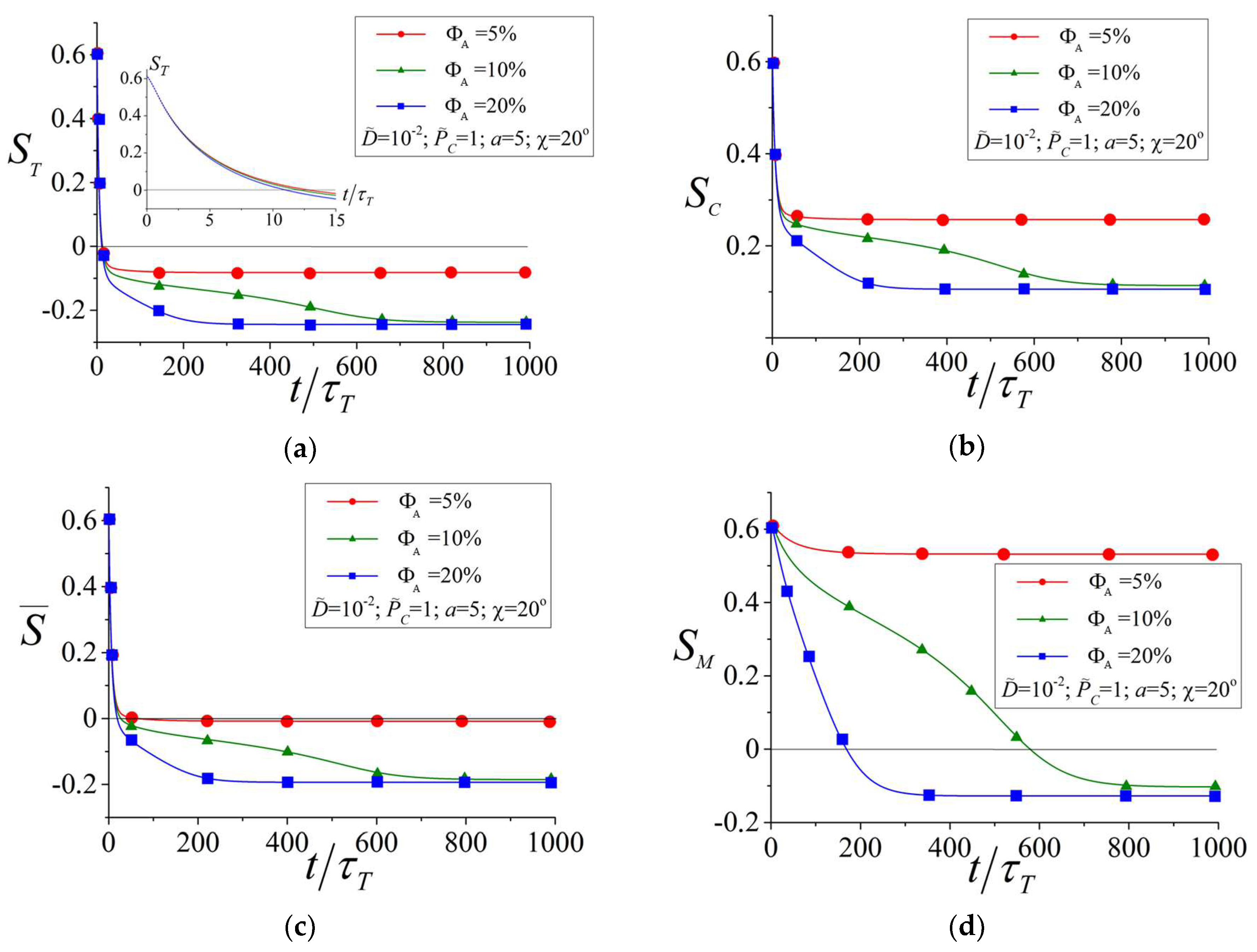
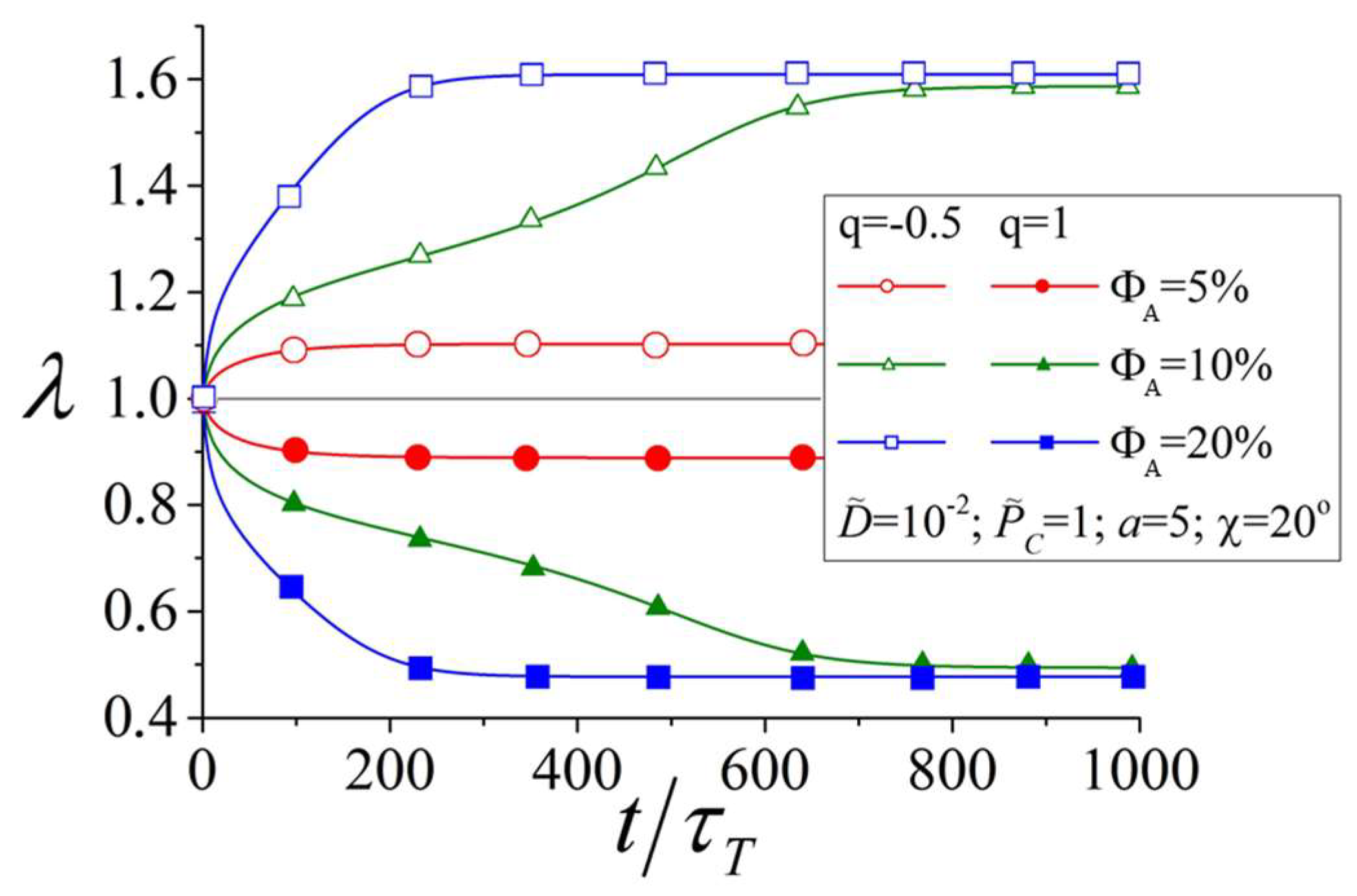
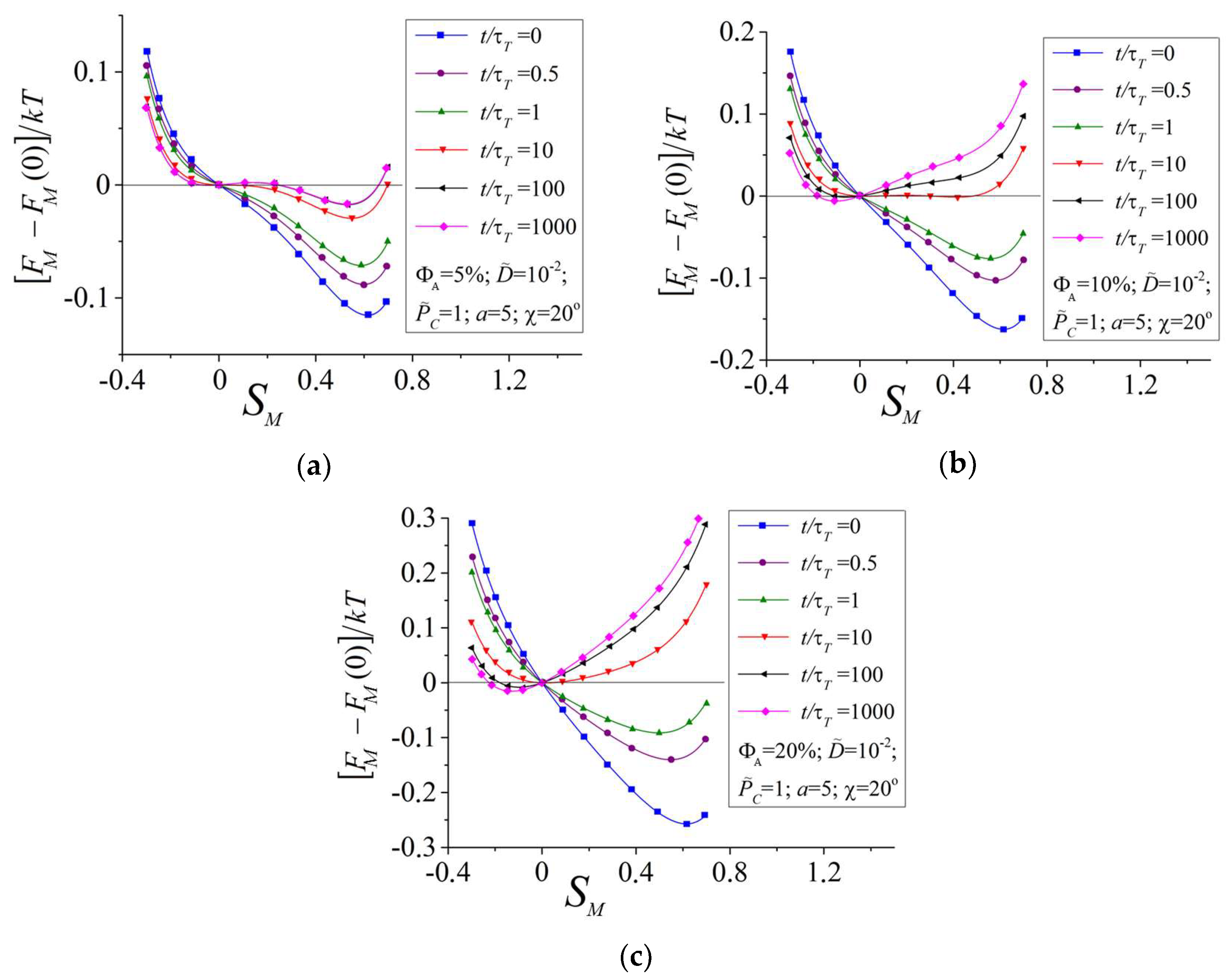
3.1. Influence of the Amount of Azobenzene Chromophores
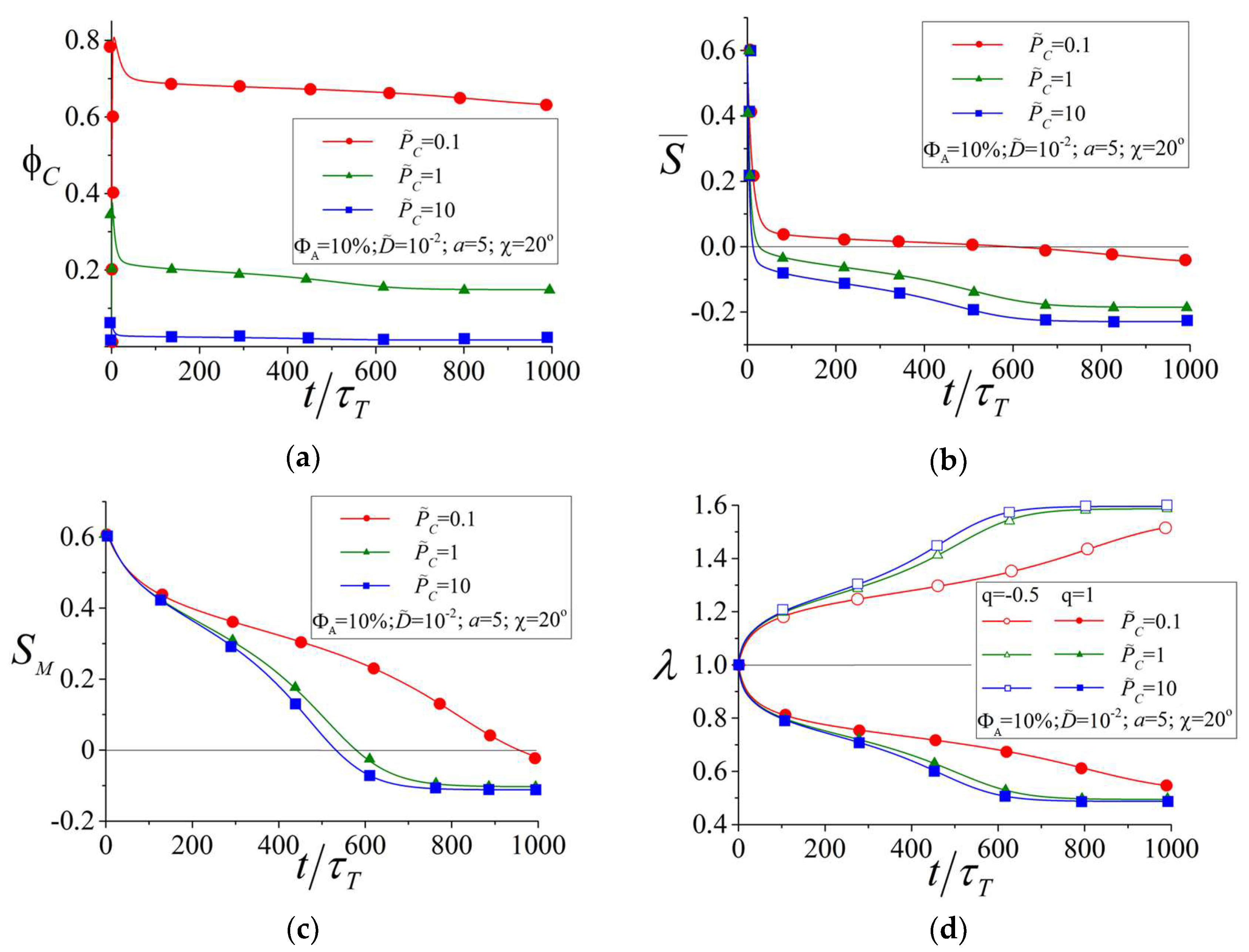
3.2. Influence of the Orientational LC Interactions between Azobenzene Chromophores and Mesogens
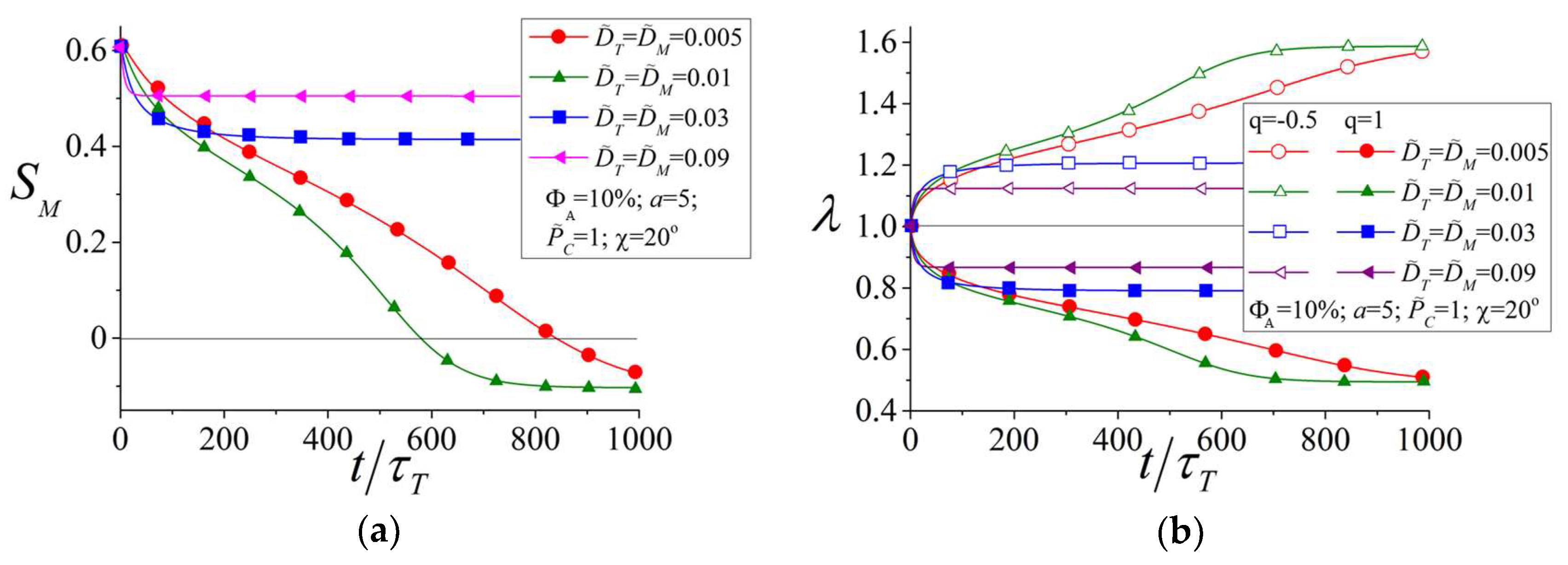
3.3. Influence of the Viscosity of the Material
3.4. Influence of the Wavelength of Light
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Kupfer, J.; Finkelmann, H. Nematic liquid single-crystal elastomers. Makromol. Chem. Rapid Commun. 1991, 12, 717–726. [Google Scholar] [CrossRef]
- Wermter, H.; Finkelmann, H. Liquid crystalline elastomers as artificial musles. e-Polymers 2001, 13, 1–13. [Google Scholar]
- Clarke, S.M.; Hotta, A.; Tajbakhsh, A.R.; Terentjev, E.M. Effect of cross-linker geometry on equilibrium thermal and mechanical properties of nematic elastomers. Phys. Rev. E 2001, 64, 061702. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.R.; Terentjev, E.M. Spontaneous thermal expansion of nematic elastomers. Eur. Phys. J. E 2001, 6, 181–188. [Google Scholar] [CrossRef]
- Warner, M.; Terentjev, E.M. Nematic elastomers—A new state of matter? Prog. Polym. Sci. 1996, 21, 853–891. [Google Scholar] [CrossRef]
- De Gennes, P.G.; Hebert, M.; Kant, R. Artificial muscles based on nematic gels. Macromol. Symp. 1997, 113, 39–49. [Google Scholar] [CrossRef]
- Warner, M.; Terentjev, E.M. Liquid Crystal Elastomers; Clarendon: Oxford, UK, 2003. [Google Scholar]
- Chigrinov, V.G.; Kozenkov, V.M.; Kwok, H.-S. Photoalignment of Liquid Crystalline Materials: Physics and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Slussarenko, S.; Murauski, A.; Du, T.; Chigrinov, V.; Marrucci, L.; Santamato, E. Tunable liquid crystal q-plates with arbitrary topological charge. Opt. Express 2011, 19, 4085–4090. [Google Scholar] [CrossRef] [PubMed]
- Tabiryan, N.; Roberts, D.; Steeves, D.; Kimball, B. New 4g optics technology extends limits to the extremes. Photonics Spectra 2017, 51, 46–50. [Google Scholar]
- Ware, T.H.; McConney, M.E.; Wie, J.J.; Tondiglia, V.P.; White, T.J. Voxelated liquid crystal elastomers. Science 2015, 347, 982–984. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Broer, D.J. Programmable and adaptive mechanics with liquid crystal polymer networks and elastomers. Nat. Mater. 2015, 14, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.J.; Kuzyk, M.G.; Neal, J.; Luchette, P.; Palffy-Muhoray, P. Experimental studies of the mechanisms of photomechanical effects in a nematic liquid crystal elastomer. J. Opt. Soc. Am. B 2011, 28, 1916–1921. [Google Scholar] [CrossRef]
- Dawson, N.J.; Kuzyk, M.G.; Neal, J.; Luchette, P.; Palffy-Muhoray, P. Modeling the mechanisms of the photomechanical response of a nematic liquid crystal elastomer. J. Opt. Soc. Am. B 2011, 28, 2134–2141. [Google Scholar] [CrossRef]
- Finkelmann, H.; Nishikawa, E.; Pereira, G.G.; Warner, M. A new opto-mechanical effect in solids. Phys. Rev. Lett. 2001, 87, 015501. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Lopez, M.; Finkelmann, H.; Palffy-Muhoray, P.; Shelley, M. Fast liquid-crystal elastomer swims into the dark. Nat. Mater. 2004, 3, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.L.; Nakano, M.; Shishido, A.; Shiono, T.; Ikeda, T. Effect of cross-linking density on photoinduced bending behavior of oriented liquid-crystalline network films containing azobenzene. Chem. Mat. 2004, 16, 1637–1643. [Google Scholar] [CrossRef]
- Sung, J.H.; Hirano, S.; Tsutsumi, O.; Kanazawa, A.; Shiono, T.; Ikeda, T. Dynamics of photochemical phase transition of guest/host liquid crystals with an azobenzene derivative as a photoresponsive chromophore. Chem. Mater. 2002, 14, 385–391. [Google Scholar] [CrossRef]
- Iamsaard, S.; Asshoff, S.J.; Matt, B.; Kudernac, T.; Cornelissen, J.; Fletcher, S.P.; Katsonis, N. Conversion of light into macroscopic helical motion. Nat. Chem. 2014, 6, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Q.; Broer, D.J. Self-assembled dynamic 3d fingerprints in liquid-crystal coatings towards controllable friction and adhesion. Angew. Chem. Int. Ed. 2014, 53, 4542–4546. [Google Scholar] [CrossRef] [PubMed]
- Wani, O.M.; Zeng, H.; Priimagi, A. A light-driven artificial flytrap. Nat. Commun. 2017, 8, 15546. [Google Scholar] [CrossRef] [PubMed]
- Gelebart, A.H.; Mulder, D.J.; Varga, M.; Konya, A.; Vantomme, G.; Meijer, E.W.; Selinger, R.L.B.; Broer, D.J. Making waves in a photoactive polymer film. Nature 2017, 546, 632. [Google Scholar] [CrossRef] [PubMed]
- Tabiryan, N.; Serak, S.; Dai, X.M.; Bunning, T. Polymer film with optically controlled form and actuation. Opt. Express 2005, 13, 7442–7448. [Google Scholar] [CrossRef] [PubMed]
- Priimagi, A.; Shimamura, A.; Kondo, M.; Hiraoka, T.; Kubo, S.; Mamiya, J.I.; Kinoshita, M.; Ikeda, T.; Shishido, A. Location of the azobenzene moieties within the cross-linked liquid-crystalline polymers can dictate the direction of photoinduced bending. ACS Macro Lett. 2012, 1, 96–99. [Google Scholar] [CrossRef]
- Lee, K.M.; White, T.J. Photochemical mechanism and photothermal considerations in the mechanical response of monodomain, azobenzene-functionalized liquid crystal polymer networks. Macromolecules 2012, 45, 7163–7170. [Google Scholar] [CrossRef]
- Wang, D.H.; Lee, K.M.; Yu, Z.N.; Koerner, H.; Vaia, R.A.; White, T.J.; Tan, L.S. Photomechanical response of glassy azobenzene polyimide networks. Macromolecules 2011, 44, 3840–3846. [Google Scholar] [CrossRef]
- Baczkowski, M.L.; Wang, D.H.; Lee, D.H.; Lee, K.M.; Smith, M.L.; White, T.J.; Tan, L.S. Photomechanical deformation of azobenzene-functionalized polyimides synthesized with bulky substituents. ACS Macro Lett. 2017, 6, 1432–1437. [Google Scholar] [CrossRef]
- White, T.J. (Ed.) Photomechnical Materails, Composites and Systems: Wireles Transduction of Light into Work; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Corbett, D.; Warner, M. Linear and nonlinear photoinduced deformations of cantilevers. Phys. Rev. Lett. 2007, 99, 174302. [Google Scholar] [CrossRef] [PubMed]
- Corbett, D.; van Oosten, C.L.; Warner, M. Nonlinear dynamics of optical absorption of intense beams. Phys. Rev. A 2008, 78, 013823. [Google Scholar] [CrossRef]
- Van Oosten, C.L.; Corbett, D.; Davies, D.; Warner, M.; Bastiaansen, C.W.M.; Broer, D.J. Bending dynamics and directionality reversal in liquid crystal network photoactuators. Macromolecules 2008, 41, 8592–8596. [Google Scholar] [CrossRef]
- Serra, F.; Terentjev, E.M. Nonlinear dynamics of absorption and photobleaching of dyes. J. Chem. Phys. 2008, 128, 224510. [Google Scholar] [CrossRef] [PubMed]
- Saphiannikova, M.; Toshchevikov, V.; Ilnytskyi, J.; Heinrich, G. Photo-induced deformation of azobenzene polymers: Theory and simulations. Proc. SPIE 2011, 8189, 818910. [Google Scholar] [CrossRef]
- Toshchevikov, V.; Saphiannikova, M.; Heinrich, G. Light-induced deformation of azobenzene elastomers: A regular cubic network model. J. Phys. Chem. B 2012, 116, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Toshchevikov, V.P.; Saphiannikova, M.; Heinrich, G. Theory of light-induced deformation of azobenzene elastomers: Influence of network structure. J. Chem. Phys. 2012, 137, 024903. [Google Scholar] [CrossRef] [PubMed]
- Toshchevikov, V.; Saphiannikova, M.; Heinrich, G. Effects of the liquid-crystalline order on the light-induced deformation of azobenzene elastomers. Proc. SPIE 2012, 8545, 854507. [Google Scholar] [CrossRef]
- Toshchevikov, V.; Saphiannikova, M. Theory of light-induced deformation of azobenzene elastomers: Effects of the liquid-crystalline interactions and biaxiality. J. Phys. Chem. B 2014, 118, 12297–12309. [Google Scholar] [CrossRef] [PubMed]
- Toshchevikov, V.; Petrova, T.; Saphiannikova, M. Light-induced deformation of liquid crystalline polymer networks containing azobenzene chromophores. Proc. SPIE 2015, 9565, 956504. [Google Scholar] [CrossRef]
- Petrova, T.; Toshchevikov, V.; Saphiannikova, M. Light-induced deformation of polymer networks containing azobenzene chromophores and liquid crystalline mesogens. Soft Matter 2015, 11, 3412–3423. [Google Scholar] [CrossRef] [PubMed]
- Saphiannikova, M.; Toshchevikov, V. Optical deformations of azobenzene polymers: Orientation approach vs. Photofluidization concept. J. Soc. Inf. Disp. 2015, 23, 146–153. [Google Scholar] [CrossRef]
- Toshchevikov, V.; Ilnytskyi, J.; Saphiannikova, M. Photoisomerization kinetics and mechanical stress in azobenzene-containing materials. J. Phys. Chem. Lett. 2017, 8, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Toshchevikov, V.; Petrova, T.; Saphiannikova, M. Kinetics of light-induced ordering and deformation in lc azobenzene-containing materials. Soft Matter 2017, 13, 2823–2835. [Google Scholar] [CrossRef] [PubMed]
- Mechau, N.; Saphiannikova, M.; Neher, D. Dielectric and mechanical properties of azobenzene polymer layers under visible and ultraviolet irradiation. Macromolecules 2005, 38, 3894–3902. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The Physics of Rubber Elasticity, 3rd ed.; Clarendon Press: Oxford, UK, 2005. [Google Scholar]
- Toshchevikov, V.P.; Gotlib, Y.Y. Shear dynamic modulus of nematic elastomers: Modified rouse model. Macromolecules 2009, 42, 3417–3429. [Google Scholar] [CrossRef]
- Toshchevikov, V.P.; Heinrich, G.; Gotlib, Y.Y. Shear dynamic moduli of stretched polymer chains and networks: Modified rouse model. Macromol. Theory Simul. 2010, 19, 195–209. [Google Scholar] [CrossRef]
- Weigert, F. A new effect of the rays in light-sensitive layers. Verh. Dtsch. Phys. Ges. 1919, 21, 479–491. [Google Scholar]
- Dumont, M.; El Osman, A. On spontaneous and photoinduced orientational mobility of dye molecules in polymers. Chem. Phys. 1999, 245, 437–462. [Google Scholar] [CrossRef]
- Radosz, W.; Pawlik, G.; Mitus, A.C. Complex dynamics of photo-switchable guest molecules in all-optical poling close to the glass transition: Kinetic monte carlo modeling. J. Phys. Chem. B 2018, 122, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, G.; Mitus, A.C.; Miniewicz, A.; Kajzar, F. Kinetics of diffraction gratings formation in a polymer matrix containing azobenzene chromophores: Experiments and monte carlo simulations. J. Chem. Phys. 2003, 119, 6789–6801. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University Press: Oxford, UK, 1988; p. 402. [Google Scholar]
- Brochard, F.; Jouffroy, J.; Levinson, P. Phase-diagrams of mesomorphic mixtures. J. Phys. 1984, 45, 1125–1136. [Google Scholar] [CrossRef]
- De Gennes, P.G.; Prost, J. The Physics of Liquid Crystals, 2nd ed.; Clarendon Press: Oxford, UK, 1995; p. 597. [Google Scholar]
- Chiu, H.W.; Kyu, T. Equilibrium phase-behavior of nematic mixtures. J. Chem. Phys. 1995, 103, 7471–7481. [Google Scholar] [CrossRef]
- Gotlib, Y.Y.; Torchinskii, I.A.; Toshchevikov, V.P. Effects of nematic ordering on the relaxation spectrum of a polymer network with included rod-like particles: Mean-field approximation. Macromol. Theory Simul. 2004, 13, 303–317. [Google Scholar] [CrossRef]
- Saphiannikova, M.; Radtchenko, I.; Sukhorukov, G.; Shchukin, D.; Yakimansky, A.; Ilnytskyi, J. Molecular-dynamics simulations and x-ray analysis of dye precipitates in the polyelectrolyte microcapsules. J. Chem. Phys. 2003, 118, 9007–9014. [Google Scholar] [CrossRef]
- Lee, K.M.; Tabiryan, N.V.; Bunning, T.J.; White, T.J. Photomechanical mechanism and structure-property considerations in the generation of photomechanical work in glassy, azobenzene liquid crystal polymer networks. J. Mater. Chem. 2012, 22, 691–698. [Google Scholar] [CrossRef]
- Hogan, P.M.; Tajbakhsh, A.R.; Terentjev, E.M. Uv manipulation of order and macroscopic shape in nematic elastomers. Phys. Rev. E 2002, 65, 041720. [Google Scholar] [CrossRef] [PubMed]
- Cviklinski, J.; Tajbakhsh, A.R.; Terentjev, E.M. Uv isomerisation in nematic elastomers as a route to photo-mechanical transducer. Eur. Phys. J. E 2002, 9, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Keller, P.; Li, B.; Wang, X.G.; Brunet, M. Light-driven side-on nematic elastomer actuators. Adv. Mater. 2003, 15, 569–572. [Google Scholar] [CrossRef]
- Ikeda, T.; Nakano, M.; Yu, Y.L.; Tsutsumi, O.; Kanazawa, A. Anisotropic bending and unbending behavior of azobenzene liquid-crystalline gels by light exposure. Adv. Mater. 2003, 15, 201–205. [Google Scholar] [CrossRef]
- Yu, Y.L.; Nakano, M.; Ikeda, T. Directed bending of a polymer film by light—Miniaturizing a simple photomechanical system could expand its range of applications. Nature 2003, 425, 145. [Google Scholar] [CrossRef] [PubMed]
- Ryabchun, A.; Bobrovsky, A.; Stumpe, J.; Shibaev, V. Novel generation of liquid crystalline photo-actuators based on stretched porous polyethylene films. Macromol. Rapid Commun. 2012, 33, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.G. The Structure and Rheology of Complex Fluids; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Tiberio, G.; Muccioli, L.; Berardi, R.; Zannoni, C. How does the trans-cis photoisomerization of azobenzene take place in organic solvents? ChemPhysChem 2010, 11, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Veer, P.U.; Pietsch, U.; Rochon, P.L.; Saphiannikova, M. Temperature dependent analysis of grating formation on azobenzene polymer films. Mol. Cryst. Liquid Cryst. 2008, 486, 1108–1120. [Google Scholar] [CrossRef]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toshchevikov, V.; Petrova, T.; Saphiannikova, M. Kinetics of Ordering and Deformation in Photosensitive Azobenzene LC Networks. Polymers 2018, 10, 531. https://doi.org/10.3390/polym10050531
Toshchevikov V, Petrova T, Saphiannikova M. Kinetics of Ordering and Deformation in Photosensitive Azobenzene LC Networks. Polymers. 2018; 10(5):531. https://doi.org/10.3390/polym10050531
Chicago/Turabian StyleToshchevikov, Vladimir, Tatiana Petrova, and Marina Saphiannikova. 2018. "Kinetics of Ordering and Deformation in Photosensitive Azobenzene LC Networks" Polymers 10, no. 5: 531. https://doi.org/10.3390/polym10050531
APA StyleToshchevikov, V., Petrova, T., & Saphiannikova, M. (2018). Kinetics of Ordering and Deformation in Photosensitive Azobenzene LC Networks. Polymers, 10(5), 531. https://doi.org/10.3390/polym10050531