Negatively Charged Porous Thin Film from ABA Triblock Copolymer Assembly




Abstract
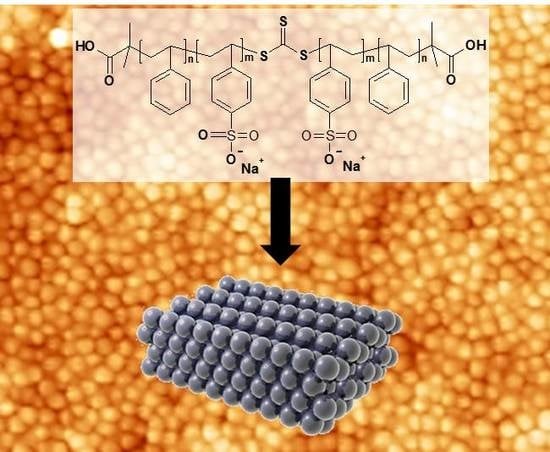
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Analytical Techniques
3. Results
3.1. Synthesis of PS10K-PNaSS10K-PS10K
3.2. Block Copolymer Micelle Preparation
3.3. Film Formation and AFM Observation
3.4. SEM and Film Thickness
3.5. Water Permeation Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Upadhyaya, L.; Semsarilar, M.; Nehache, S.; Deratani, A.; Quemener, D. Filtration membranes from self-assembled block copolymers—A review on recent progress. Eur. Phys. J. Spec. Top. 2015, 224, 1883–1897. [Google Scholar] [CrossRef]
- Schacher, F.H.; Rupar, P.A.; Manners, I. Functional block copolymers: Nanostructured materials with emerging applications. Angew. Chem. Int. Ed. 2012, 51, 7898–7921. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Patra, S.K.; Hamley, I.W.; Manners, I.; Faul, C.F.J. Tetragonal and helical morphologies from polyferrocenylsilane block polyelectrolytes via ionic self-assembly. J. Am. Chem. Soc. 2013, 135, 2455–2458. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Dobbins, D.J.; Dai, Y.; Kabb, C.P.; Wu, S.; Alfurhood, J.A.; Rinaldi, C.; Sumerlin, B.S. Radical Departure: Thermally-Triggered Degradation of Azo-Containing Poly(β-thioester)s. ACS Macro Lett. 2016, 5, 688–693. [Google Scholar] [CrossRef]
- Ramanathan, M.; Kilbey, S.M.; Ji, Q.M.; Hill, J.P.; Ariga, K. Materials self-assembly and fabrication in confined spaces. J. Mater. Chem. 2012, 22, 10389–10405. [Google Scholar] [CrossRef]
- Tan, J.; Sun, H.; Yu, M.; Sumerlin, B.S.; Zhang, L. Photo-PISA: Shedding Light on Polymerization-Induced Self-Assembly. ACS Macro Lett. 2015, 4, 1249–1253. [Google Scholar] [CrossRef]
- Ren, L.X.; Hardy, C.G.; Tang, C.B. Synthesis and solution self-assembly of side-chain cobaltocenium-containing block copolymers. J. Am. Chem. Soc. 2010, 132, 8874–8875. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.J.; Jiang, M.; Peng, H.S.; Chen, D.Y.; Hong, Y. pH-dependent self-assembly: Micellization and micelle-hollow-sphere transition of cellulose-based copolymers. Angew. Chem. Int. Ed. 2003, 42, 1516–1519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.K.; Jiang, M.; Chen, D.Y. DNA/polymeric micelle self-assembly mimicking chromatin compaction. Angew. Chem. Int. Ed. 2012, 51, 8744–8747. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.G.; Audus, D.J.; Klinger, D.; Krogstad, D.V.; Kim, B.J.; Cameron, A.; Kim, S.W.; Delaney, K.T.; Hur, S.M.; Killops, K.L.; et al. Striped, ellipsoidal particles by controlled assembly of diblock copolymers. J. Am. Chem. Soc. 2013, 135, 6649–6657. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.G.; Kramer, E.J.; Hawker, C.J. Controlled supramolecular assembly of micelle-like gold nanoparticles in PS-b-P2VP diblock copolymers via hydrogen bonding. J. Am. Chem. Soc. 2011, 133, 16986–16996. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Fullhart, P.; Mirkin, C.A. Reversible and chemically programmable micelle assembly with DNA block-copolymer amphiphiles. Nano Lett. 2004, 4, 1055–1058. [Google Scholar] [CrossRef]
- Betthausen, E.; Dulle, M.; Hanske, C.; Muller, M.; Fery, A.; Forster, S.; Schacher, F.H.; Muller, A.H.E. Nanoporous sheets and cylinders via bulk templating of triblock terpolymer/homopolymer blends. Macromolecules 2014, 47, 6289–6301. [Google Scholar] [CrossRef]
- Darling, S.B. Directing the self-assembly of block copolymers. Prog. Polym. Sci. 2007, 32, 1152–1204. [Google Scholar] [CrossRef]
- Kim, J.K.; Yang, S.Y.; Lee, Y.; Kim, Y. Functional nanomaterials based on block copolymer self-assembly. Prog. Polym. Sci. 2010, 35, 1325–1349. [Google Scholar] [CrossRef]
- Olson, D.A.; Chen, L.; Hillmyer, M.A. Templating nanoporous polymers with ordered block copolymers. Chem. Mater. 2008, 20, 869–890. [Google Scholar] [CrossRef]
- Bates, F.S. Polymer-polymer phase-behavior. Science 1991, 251, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.; Amendt, M.A.; Hillmyer, M.A. Cross-linked nanoporous materials from reactive and multifunctional block polymers. Macromolecules 2011, 44, 9310–9318. [Google Scholar] [CrossRef]
- Seo, M.; Hillmyer, M.A. Reticulated nanoporous polymers by controlled polymerization-induced microphase separation. Science 2012, 336, 1422–1425. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Xu, F.; Sun, B.; Fu, R.W.; He, H.K.; Matyjaszewski, K. Design and preparation of porous polymers. Chem. Rev. 2012, 112, 3959–4015. [Google Scholar] [CrossRef] [PubMed]
- Peinemann, K.V.; Abetz, V.; Simon, P.F.W. Asymmetric superstructure formed in a block copolymer via phase separation. Nat. Mater. 2007, 6, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Marques, D.S.; Vainio, U.; Chaparro, N.M.; Calo, V.M.; Bezahd, A.R.; Pitera, J.W.; Peinemann, K.V.; Nunes, S.P. Self-assembly in casting solutions of block copolymer membranes. Soft Matter 2013, 9, 5557–5564. [Google Scholar] [CrossRef]
- Guo, L.M.; Wang, Y. Nanoslitting of phase-separated block copolymers by solvent swelling for membranes with ultrahigh flux and sharp selectivity. Chem. Commun. 2014, 50, 12022–12025. [Google Scholar] [CrossRef] [PubMed]
- Schacher, F.; Ulbricht, M.; Müller, A.H.E. Self-supporting, double stimuli-responsive porous membranes from polystyrene-block-poly(N,N-dimethylaminoethyl methacrylate) diblock copolymers. Adv. Funct. Mater. 2009, 19, 1040–1045. [Google Scholar] [CrossRef]
- Yang, S.Y.; Yang, J.A.; Kim, E.S.; Jeon, G.; Oh, E.J.; Choi, K.Y.; Hahn, S.K.; Kim, J.K. Single-file diffusion of protein drugs through cylindrical nanochannels. ACS Nano 2010, 4, 3817–3822. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.A.; Hillmyer, M.A. Nanoporous membranes derived from block copolymers: From drug delivery to water filtration. ACS Nano 2010, 4, 3548–3553. [Google Scholar] [CrossRef] [PubMed]
- Abetz, V. Isoporous Block Copolymer Membranes. Macromol. Rapid Commun. 2015, 36, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.P. Block Copolymer Membranes for Aqueous Solution Applications. Macromolecules 2016, 49, 2905–2916. [Google Scholar] [CrossRef] [Green Version]
- Nunes, S.P.; Sougrat, R.; Hooghan, B.; Anjum, D.H.; Behzad, A.R.; Zhao, L.; Pradeep, N.; Pinnau, I.; Vainio, U.; Peinemann, K.V. Ultraporous Films with Uniform Nanochannels by Block Copolymer Micelles Assembly. Macromolecules 2015, 48, 6153–6159. [Google Scholar] [CrossRef]
- Mulvenna, R.A.; Weidman, J.L.; Jing, B.; Pople, J.A.; Zhu, Y.; Boudouris, B.W.; Phillip, W.A. Tunable nanoporous membranes with chemically-tailored pore walls from triblock polymer templates. J. Membr. Sci. 2014, 470, 246–256. [Google Scholar] [CrossRef]
- Shevate, R.; Karunakaran, M.; Kumar, M.; Peinemann, K.V. Polyanionic pH-responsive polystyrene-b-poly(4-vinyl pyridine-N-oxide) isoporous membranes. J. Membr. Sci. 2016, 501, 161–168. [Google Scholar] [CrossRef]
- Menzel, C.; Cambon, A.; Yeates, S.G. Double emulsion template suspension polymerisation: Towards the synthesis of polyelectrolyte core porous hydrophobic shell particles for environmental applications. J. Mater. Chem. A 2013, 1, 12553–12559. [Google Scholar] [CrossRef]
- Hickner, M.A.; Ghassemi, H.; Kim, Y.S.; Einsla, B.R.; McGrath, J.E. Alternative polymer systems for proton exchange membranes (PEMs). Chem. Rev. 2004, 104, 4587–4611. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T. Progress in membrane science and technology for seawater desalination—A review. Desalination 2001, 134, 47–54. [Google Scholar] [CrossRef]
- Raviv, U.; Giasson, S.; Kampf, N.; Gohy, J.F.; Jerome, R.; Klein, J. Lubrication by charged polymers. Nature 2003, 425, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Wittemann, A.; Haupt, B.; Ballauff, M. Adsorption of proteins on spherical polyelectrolyte brushes in aqueous solution. Phys. Chem. Chem. Phys. 2003, 5, 1671–1677. [Google Scholar] [CrossRef] [Green Version]
- Schallon, A.; Jerome, V.; Walther, A.; Synatschke, C.V.; Muller, A.H.E.; Freitag, R. Performance of three PDMAEMA-based polycation architectures as gene delivery agents in comparison to linear and branched PEI. React. Funct. Polym. 2010, 70, 1–10. [Google Scholar] [CrossRef]
- McCullough, P.A.; Beaver, T.M.; Bennett-Guerrero, E.; Emmett, M.; Fonarow, G.C.; Goyal, A.; Herzog, C.A.; Kosiborod, M.; Palmer, B.F. Acute and chronic cardiovascular effects of hyperkalemia: New insights into prevention and clinical management. Rev. Cardiovasc. Med. 2014, 15, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Spinner, I.H.; Ciric, J.; Graydon, W.F. Preparation of ion-exchange resins. Can. J. Chem. 1954, 32, 143–152. [Google Scholar] [CrossRef]
- Krol, J.J.; Wessling, M.; Strathmann, H. Concentration polarization with monopolar ion exchange membranes: Current-voltage curves and water dissociation. J. Membr. Sci. 1999, 162, 145–154. [Google Scholar] [CrossRef]
- Sata, T.; Yoshida, T.; Matsusaki, K. Transport properties of phosphonic acid and sulfonic acid cation exchange membranes. J. Membr. Sci. 1996, 120, 101–110. [Google Scholar] [CrossRef]
- Smitha, B.; Sridhar, S.; Khan, A.A. Solid polymer electrolyte membranes for fuel cell applications—A review. J. Membr. Sci. 2005, 259, 10–26. [Google Scholar] [CrossRef]
- Lai, J.T.; Filla, D.; Shea, R. Functional polymers from novel carboxyl-terminated trithiocarbonates as highly efficient RAFT agents. Macromolecules 2002, 35, 6754–6756. [Google Scholar] [CrossRef]
- Nehache, S.; Semsarilar, M.; In, M.; Dieudonne-George, P.; Lai-Kee-Him, J.; Bron, P.; Bouyer, D.; Deratani, A.; Quemener, D. Self-assembly of PS-PNaSS-PS triblock copolymers from solution to the solid state. Polym. Chem. 2017, 8, 3357–3363. [Google Scholar] [CrossRef]
- Nehache, S.; Semsarilar, M.; Deratani, A.; In, M.; Dieudonné-George, P.; Lai Kee Him, J.; Bron, P.; Quémener, D. Nano-porous structures via self-assembly of amphiphilic triblock copolymers: Influence of solvent and molecular weight. Polym. Chem. 2018, 9, 193–202. [Google Scholar] [CrossRef]
- Tyagi, P.; Deratani, A.; Bouyer, D.; Cot, D.; Gence, V.; Barboiu, M.; Phan, T.N.T.; Bertin, D.; Gigmes, D.; Quemener, D. Dynamic interactive membranes with pressure-driven tunable porosity and self-healing ability. Angew. Chem. Int. Ed. 2012, 51, 7166–7170. [Google Scholar] [CrossRef] [PubMed]
- Remigy, J.-C.; Desclaux, S. Filtration membranaire (OI, NF, UF)-Présentation des membranes et modules. Tech. L’Ing. W 2007, 4090, 20. [Google Scholar]
- Zhu, Z.B.; Wan, W.; Deng, Z.P.; Ge, Z.Y.; Huo, L.H.; Zhao, H.; Gao, S. Structure modulations in luminescent alkaline earth metal-sulfonate complexes constructed from dihydroxyl-1,5-benzenedisulfonic acid: Influences of metal cations, coordination modes and pH value. CrystEngComm 2012, 14, 6675–6688. [Google Scholar] [CrossRef]
- Wang, G.; Lee, K.H.; Lee, W.H.; Shin, D.W.; Kang, N.R.; Cho, D.H.; Hwang, D.S.; Zhuang, Y.; Lee, Y.M.; Guiver, M.D. Durable sulfonated poly(benzothiazole-co-benzimidazole) proton exchange membranes. Macromolecules 2014, 47, 6355–6364. [Google Scholar] [CrossRef]
- He, Y.K.; Wang, J.T.; Zhang, H.Q.; Zhang, T.; Zhang, B.; Cao, S.K.; Liu, J.D. Polydopamine-modified graphene oxide nanocomposite membrane for proton exchange membrane fuel cell under anhydrous conditions. J. Mater. Chem. A 2014, 2, 9548–9558. [Google Scholar] [CrossRef]
- Henkensmeier, D.; Gubler, L. Shape memory effect in radiation grafted ion exchange membranes. J. Mater. Chem. A 2014, 2, 9482–9485. [Google Scholar] [CrossRef]
- Ramaswamy, P.; Matsuda, R.; Kosaka, W.; Akiyama, G.; Jeon, H.J.; Kitagawa, S. Highly proton conductive nanoporous coordination polymers with sulfonic acid groups on the pore surface. Chem. Commun. 2014, 50, 1144–1146. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nehache, S.; Semsarilar, M.; Deratani, A.; Quemener, D. Negatively Charged Porous Thin Film from ABA Triblock Copolymer Assembly. Polymers 2018, 10, 733. https://doi.org/10.3390/polym10070733
Nehache S, Semsarilar M, Deratani A, Quemener D. Negatively Charged Porous Thin Film from ABA Triblock Copolymer Assembly. Polymers. 2018; 10(7):733. https://doi.org/10.3390/polym10070733
Chicago/Turabian StyleNehache, Sabrina, Mona Semsarilar, André Deratani, and Damien Quemener. 2018. "Negatively Charged Porous Thin Film from ABA Triblock Copolymer Assembly" Polymers 10, no. 7: 733. https://doi.org/10.3390/polym10070733
APA StyleNehache, S., Semsarilar, M., Deratani, A., & Quemener, D. (2018). Negatively Charged Porous Thin Film from ABA Triblock Copolymer Assembly. Polymers, 10(7), 733. https://doi.org/10.3390/polym10070733