Confirmation of Bioinformatics Predictions of the Structural Domains in Honeybee Silk

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
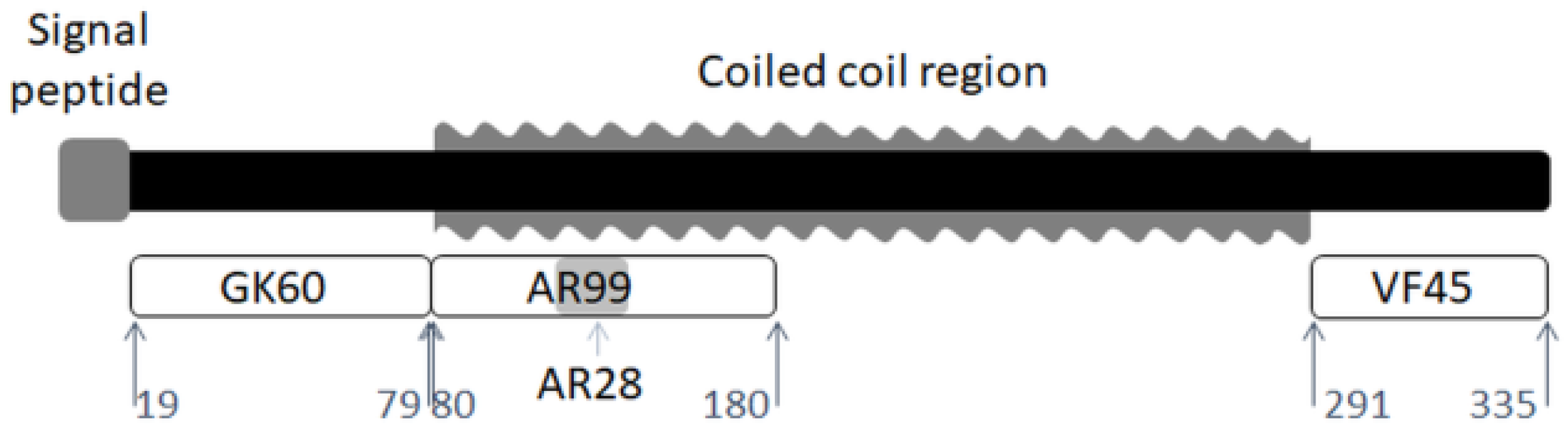
2.1. Samples
2.2. Fabrication Methods
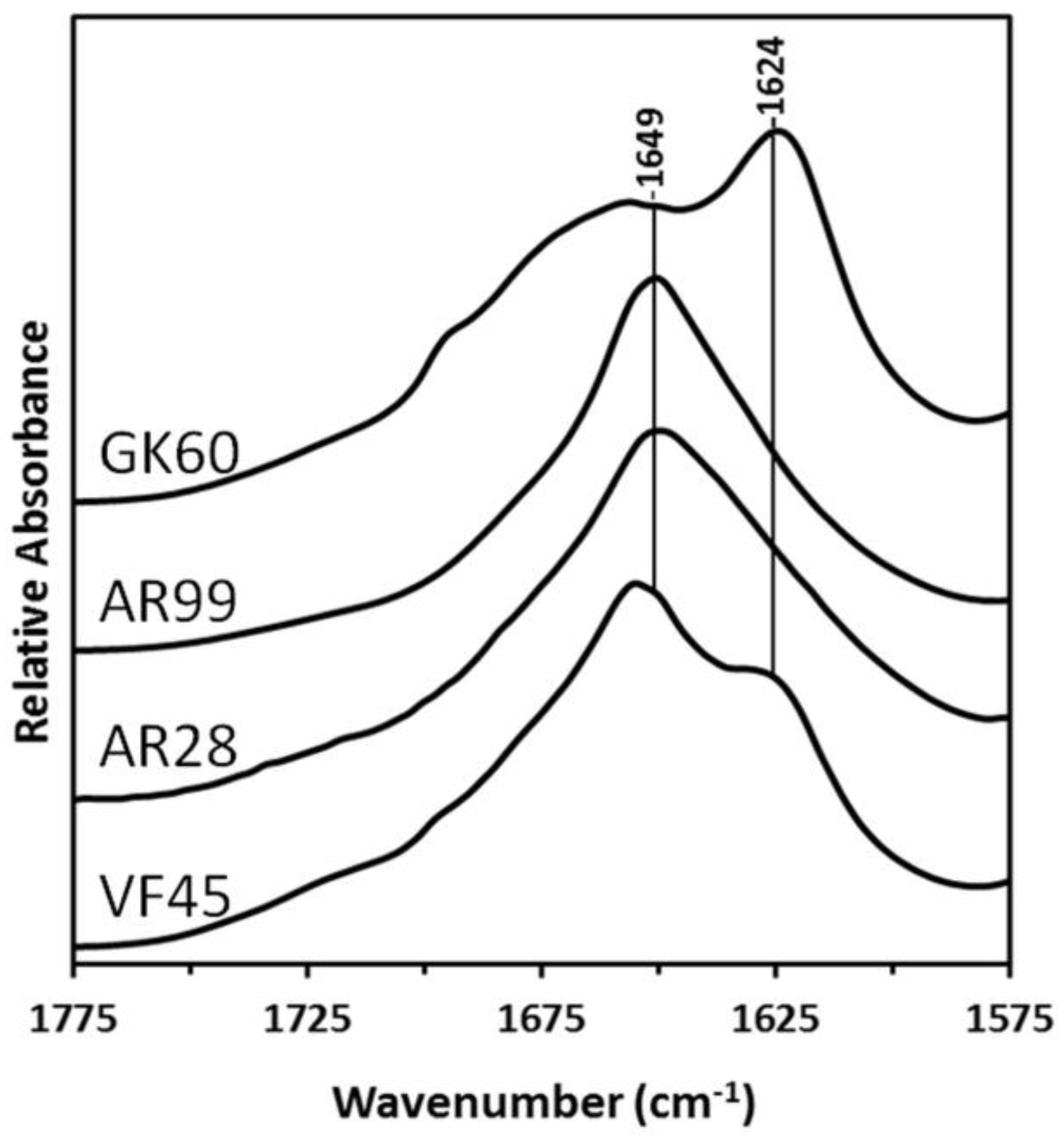
2.3. Fourier Transform Infrared (FTIR) Spectroscopy
2.4. Circular Dichroism (CD) Spectroscopy
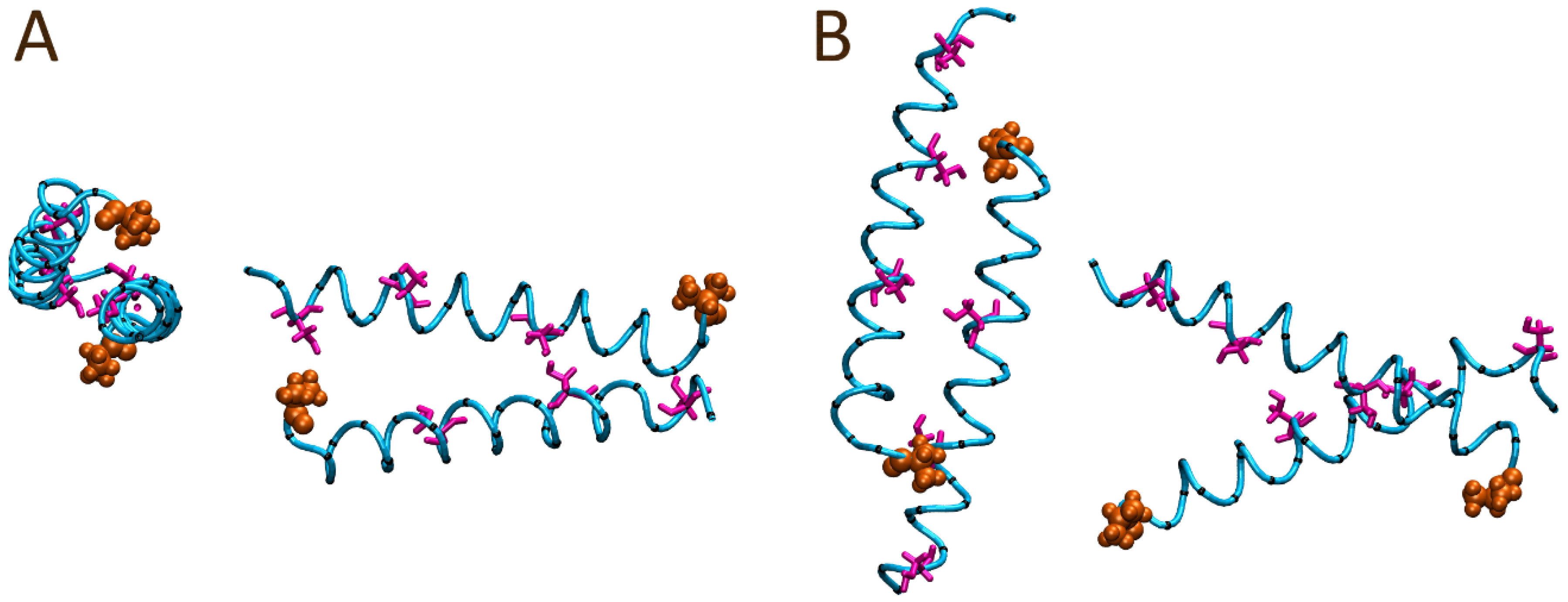
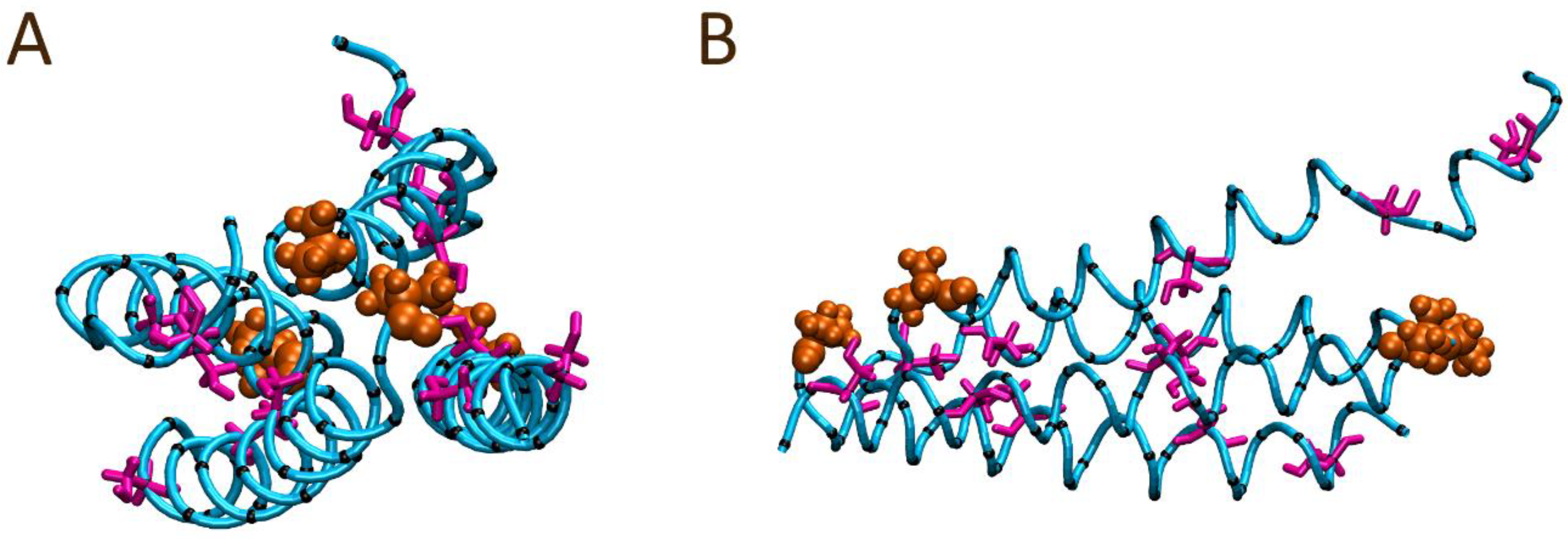
2.5. Molecular Dynamics (MD) Simulations
3. Results
3.1. Structure of Peptides in the Solid-State
3.2. Structure of Peptides in Solution
3.3. The Role of the N- and C-Terminal Regions in Material Formation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hepburn, H.R.; Kurstjens, S.P. The combs of honeybees as composite materials. Apidologie 1988, 19, 25–36. [Google Scholar] [CrossRef]
- Silva-Zacarin, E.C.M.; Silva De Moraes, R.L.M.; Taboga, S.R. Silk formation mechanisms in the larval salivary glands of Apis mellifera (Hymenoptera:Apidae). J. Biosci. 2003, 28, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Kameda, T.; Tamada, Y. Variable-temperature 13C solid-state NMR study of the molecular structure of honeybee wax and silk. Int. J. Biol. Macromol. 2009, 44, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, T.D.; Campbell, P.M.; Weisman, S.; Trueman, H.E.; Sriskantha, A.; Wanjura, W.J.; Haritos, V.S. A highly divergent gene cluster in honey bees encodes a novel silk family. Genome Res. 2006, 16, 1414–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.M.; Weisman, S.; Trueman, H.E.; Sriskantha, A.; Wanjura, W.J.; Haritos, V.S. Cross-linking in the silks of bees, ants and hornets. Insect Biochem. Mol. Biol. 2014, 48, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Kambe, Y.; Sutherland, T.D.; Kameda, T. Recombinant production and film properties of full-length hornet silk proteins. ACTA Biomater. 2014, 10, 3590–3598. [Google Scholar] [CrossRef] [PubMed]
- Maitip, J.; Trueman, H.E.; Kaehler, B.D.; Huttley, G.A.; Chantawannakul, P.; Sutherland, T.D. Folding behaviour of four silks of giant honeybee reflects the evolutionary conservation of aculeate silk proteins. Insect Biochem. Mol. Biol. 2015, 48, 40–50. [Google Scholar]
- Weisman, S.; Haritos, V.S.; Church, J.S.; Huson, M.G.; Mudie, S.T.; Rodgers, A.J.; Dumsdayd, G.J.; Sutherlanda, T.D. Honeybee silk: Recombinant protein production, assembly and fiber spinning. Biomaterials 2010, 31, 2695–2700. [Google Scholar] [CrossRef] [PubMed]
- Huson, M.G.; Church, J.S.; Poole, J.M.; Weisman, S.; Sriskantha, S.; Warden, A.C.; Ramshaw, J.A.M.; Sutherland, T.D. Structural and physical changes of honeybee silk materials induced by heating or by immersion in aqueous methanol solutions. PLoS ONE 2012, 7, e52308. [Google Scholar] [CrossRef] [PubMed]
- Poole, J.M.; Church, J.S.; Woodhead, A.L.; Huson, M.G.; Sriskantha, A.; Kyratzis, I.L.; Sutherland, T.D. Continuous production of flexible fibers from transgenically-produced honeybee silk proteins. Macromol. Biosci. 2013, 13, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Rapson, T.D.; Church, J.S.; Trueman, H.E.; Dacres, H.; Sutherland, T.D.; Trowell, S.C. Micromolar biosensing of nitric oxide using myoglobin immobilised in a synthetic silk film. Biosens. Bioelectron. 2014, 62, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Wittmer, C.R.; Hu, X.; Gauthier, P.C.; Weisman, S.; Kaplan, D.L.; Sutherland, T.D. Production, structure and in vitro degradation of electrospun honeybee silk nanofibres. Acta Biomater. 2011, 7, 3789–3795. [Google Scholar] [CrossRef] [PubMed]
- Horgan, C.C.; Han, Y.S.; Trueman, H.; Jackson, C.J.; Sutherland, T.D.; Rapson, T.D. Phosphorescent oxygen-sensing and singlet oxygen production by a biosynthetic silk. RSC Adv. 2016, 6, 39530–39533. [Google Scholar] [CrossRef] [Green Version]
- Rapson, T.D.; Kusuoka, R.; Butcher, J.; Musameh, M.; Dunn, C.J.; Church, J.S.; Warden, A.C.; Blanford, C.F.; Nakamura, N.; Sutherland, T.D. Bioinspired electrocatalysts for oxygen reduction using recombinant silk films. J. Mater. Chem. A 2017, 5, 10236–10243. [Google Scholar] [CrossRef] [Green Version]
- Rapson, T.D.; Sutherland, T.D.; Church, J.S.; Trueman, H.E.; Dacres, H.; Trowell, S.C. De novo engineering of solid-state metalloproteins using recombinant coiled-coil silk. ACS Biomater. Sci. Eng. 2015, 1, 1114–1120. [Google Scholar] [CrossRef]
- Delorenzi, M.; Speed, T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 2002, 18, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupas, A.; Van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Berger, B.; Wilson, D.B.; Wolf, E.; Tonchev, T.; Milla, M.; Kim, P.S. Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. USA 1995, 92, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Kim, P.S.; Berger, B. Multicoil: A program for predicting two- and three-stranded coiled coils. Protein Sci. 1997, 6, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, T.D.; Trueman, H.E.; Walker, A.A.; Weisman, S.; Campbell, P.M.; Dong, Z.; Church, J.S.; Husond, M.G.; Woodhead, A.L. Convergently-evolved structural anomalies in the coiled coil domains of insect silk proteins. J. Struct. Biol. 2014, 186, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.A.D. Coiled-coils in alpha-helix-containing proteins: Analysis of the residue types within the heptad repeat and the use of these data in the prediction of coiled coils in other proteins. Biosci. Rep. 1982, 2, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Atkins, E.D.T. A four-strand coiled-coil model for some insect fibrous proteins. J. Mol. Biol. 1967, 24, 139–141. [Google Scholar] [CrossRef]
- Fraser, R.D.; Parry, D.A. The molecular structure the silk fibers from Hymenoptera aculeate (bees, wasps, ants). J. Struct. Biol. 2015, 192, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Kameda, T.; Kojima, K.; Miyazawa, M.; Fujiwara, S. Film formation and structural characterization of silk of the hornet Vespa simillima xanthoptera Cameron. Z. Naturforsch. C 2005, 60, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Kameda, T. Hornet (Vespa) silk composed of coiled-coil proteins. Kobunshi Ronbunshu 2010, 67, 641–653. [Google Scholar] [CrossRef]
- Kameda, T.; Kojima, K.; Togawa, E.; Sezutsu, H.; Zhang, Q.; Teramoto, H.; Tamada, Y. Drawing-induced changes in morphology and mechanical properties of hornet silk gel films. Biomacromolecules 2010, 11, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Sezutzu, H.; Kajiwara, H.; Kojima, K.; Mita, K.; Tamura, T.; Tamada, Y.; Kameda, T. Identification of four major hornet silk genes with a complex of alanine-rich and serine-rich sequences in Vespa simillima xanthoptera Cameron. Biosci. Biotechnol. Biochem. 2007, 71, 2725–2734. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How robust are protein folding simulations with respect to force field parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hockney, R.W.; Goel, S.P.; Eastwood, J.W. Quiet high-resolution computer models of a plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 2002, 100, 191–198. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transistions in single-crystals—A new molecular-dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Byler, D.M.; Susi, H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 1986, 25, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Dong, A.; Huang, P.; Caughey, W.S. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry 1990, 29, 3303–3308. [Google Scholar] [CrossRef] [PubMed]
- Heimburg, T.; Schuenemann, J.; Weber, K.; Geisler, N. Specific recognition of coiled coils by infrared spectroscopy: Analysis of the three structural domains of type III Intermediate filament proteins. Biochemistry 1996, 35, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.A.; Warden, A.C.; Trueman, H.E.; Weisman, S.; Sutherland, T.D. Micellar refolding of coiled coil honeybee silk proteins. J. Mater. Chem. B 2013, 1, 3644–3651. [Google Scholar] [CrossRef]
- Zhou, N.E.; Kay, C.M.; Hodges, R.S. The net energetic contribution of interhelical electrostatic attractions to coiled coil stability. Protein Eng. 1994, 7, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Walshaw, J.; Woolfson, D.N. SOCKET: A program for identifying and analysing coiled-coil motifs within protein structures11Edited by J. Thornton. J. Mol. Biol. 2001, 307, 1427–1450. [Google Scholar] [CrossRef] [PubMed]
- Walther, D.; Eisenhaber, F.; Argos, P. Principles of Helix-Helix Packing in Proteins: The Helical Lattice Superposition Model. J. Mol. Biol. 1996, 255, 536–553. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.H.; Kim, K.H.; Jun, G.; Greenfield, N.J.; Dominguez, R.; Volkmann, N.; Cohen, C.; Hitchcock-DeGregori, S.E. Deciphering the design of the tropomyosin molecule. Proc. Natl. Acad. Sci. USA 2001, 98, 8496–8501. [Google Scholar] [CrossRef] [PubMed]
- Gernert, K.M.; Surles, M.C.; Labean, T.H.; Richardson, J.S.; Richardson, D.C. The Alacoil: A very tight, antiparallel coiled-coil of helices. Protein Sci. 1995, 4, 2252–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolfson, D.N.; Mahmoud, Z.N. More than just bare scaffolds: Towards multi-component and decorated fibrous biomaterials. Chem. Soc. Rev. 2010, 39, 3464–3479. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zheng, Q.; Liu, J.; Cheng, C.S.; Kallenbach, N.R.; Lu, M. Self-assembly of coiled coil tetramers in the 1.40 Å structure of a leucine-zipper mutant. Protein Sci. 2007, 16, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.A.; Holland, C.; Sutherland, T.D. More than one way to spin a crystallite: Multiple trajectories through liquid crystallinity to solid silk. Proc. R. Soc. B 2015, 282, 20150259. [Google Scholar] [CrossRef] [PubMed]
- Flower, N.E.; Kenchington, W. Studies on insect fibrous proteins: The larval silk of Apis, Bombus and Vespa (Hymenoptera: Aculeata). J. R. Microsc. Soc. 1967, 86, 297–310. [Google Scholar] [CrossRef]
- Sutherland, T.D.; Weisman, S.; Trueman, H.E.; Sriskantha, A.; Trueman, J.W.; Haritos, V.S. Conservation of essential design features in coiled coil silks. Mol. Biol. Evol. 2007, 24, 2424–2432. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
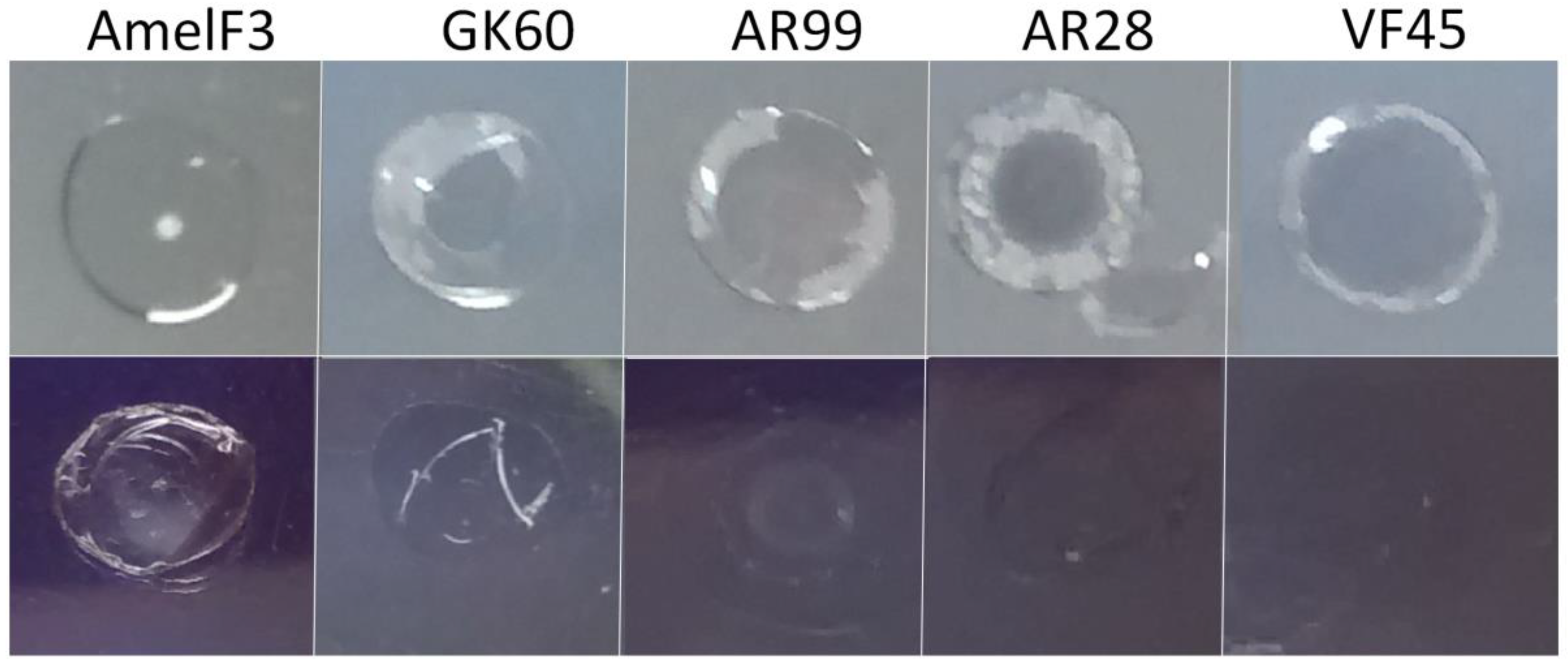{kind=link}
| Conformation (%) | GK60 (N-Terminal) | AR99 (Coiled Coil) | AR28 (Coiled Coil) | VF45 (C-Terminal) | ||||
|---|---|---|---|---|---|---|---|---|
| CF | MeOH | CF | MeOH | CF | MeOH | CF | MeOH | |
| Coiled coil | 14 | 12 | 86 | 18 | 85 | 13 | 19 | 10 |
| β-sheet | 50 | 59 | 0 | 49 | 0 | 54 | 44 | 65 |
| β-turn | 28 | 23 | 0 | 23 | 0 | 28 | 28 | 19 |
| Unordered | 9 | 6 | 14 | 9 | 15 | 5 | 10 | 6 |
| α-Helix Region | Starting Residue | Ending Residue |
|---|---|---|
| 1 | Leu6 | Lys11 |
| 2 | Leu13 | Gln19 |
| 3 | Glu21 | Ala25 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woodhead, A.L.; Church, A.T.; Rapson, T.D.; Trueman, H.E.; Church, J.S.; Sutherland, T.D. Confirmation of Bioinformatics Predictions of the Structural Domains in Honeybee Silk. Polymers 2018, 10, 776. https://doi.org/10.3390/polym10070776
Woodhead AL, Church AT, Rapson TD, Trueman HE, Church JS, Sutherland TD. Confirmation of Bioinformatics Predictions of the Structural Domains in Honeybee Silk. Polymers. 2018; 10(7):776. https://doi.org/10.3390/polym10070776
Chicago/Turabian StyleWoodhead, Andrea L., Andrew T. Church, Trevor D. Rapson, Holly E. Trueman, Jeffrey S. Church, and Tara D. Sutherland. 2018. "Confirmation of Bioinformatics Predictions of the Structural Domains in Honeybee Silk" Polymers 10, no. 7: 776. https://doi.org/10.3390/polym10070776
APA StyleWoodhead, A. L., Church, A. T., Rapson, T. D., Trueman, H. E., Church, J. S., & Sutherland, T. D. (2018). Confirmation of Bioinformatics Predictions of the Structural Domains in Honeybee Silk. Polymers, 10(7), 776. https://doi.org/10.3390/polym10070776