Oral Vaccine Delivery for Intestinal Immunity—Biological Basis, Barriers, Delivery System, and M Cell Targeting


Abstract
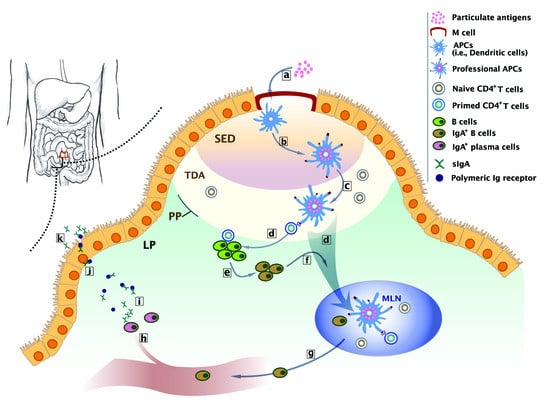
:
1. Introduction
2. Biological Basis of Intestinal Immunity
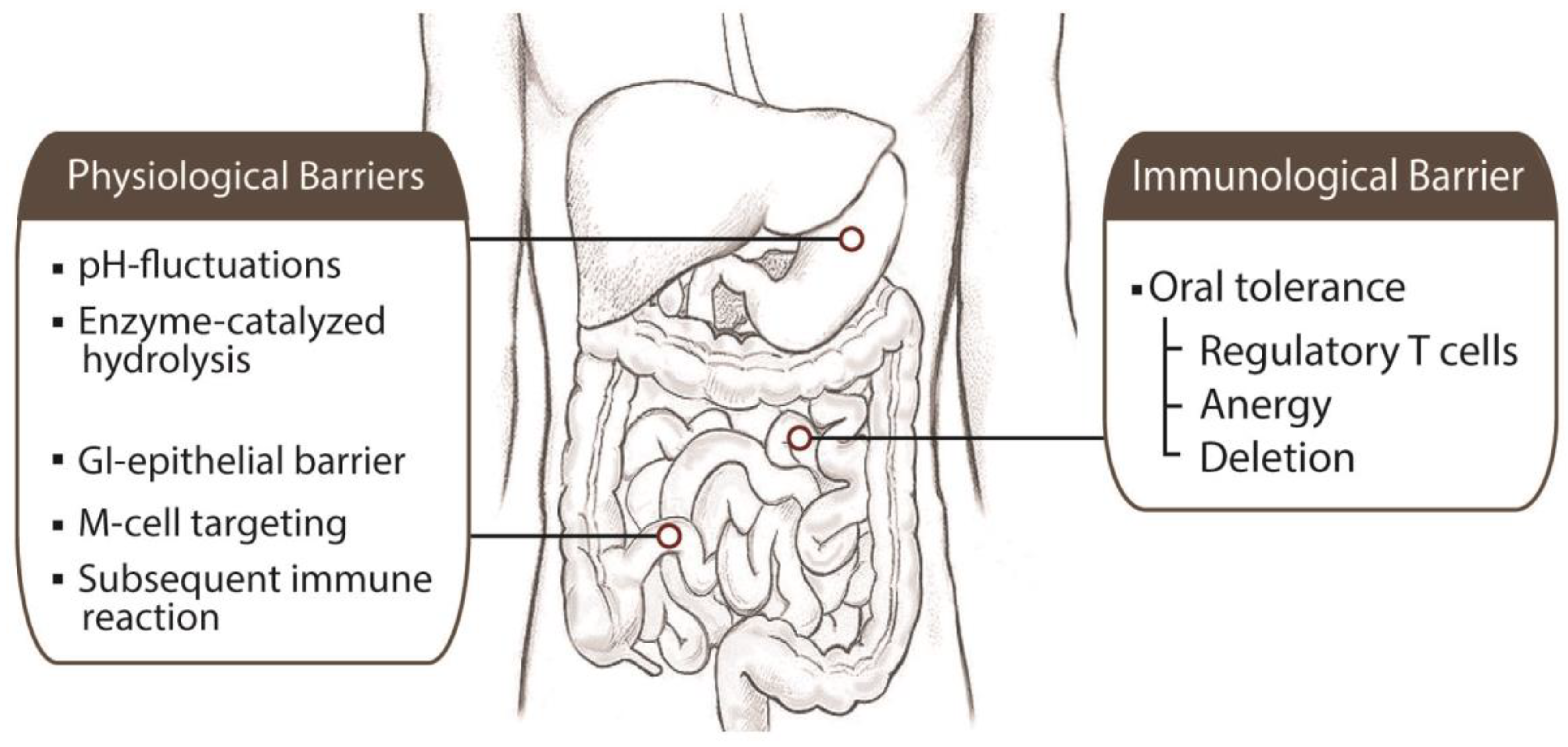
3. Barriers to Oral Vaccine Delivery
3.1. Physiological Barriers
3.2. Immunological Barrier
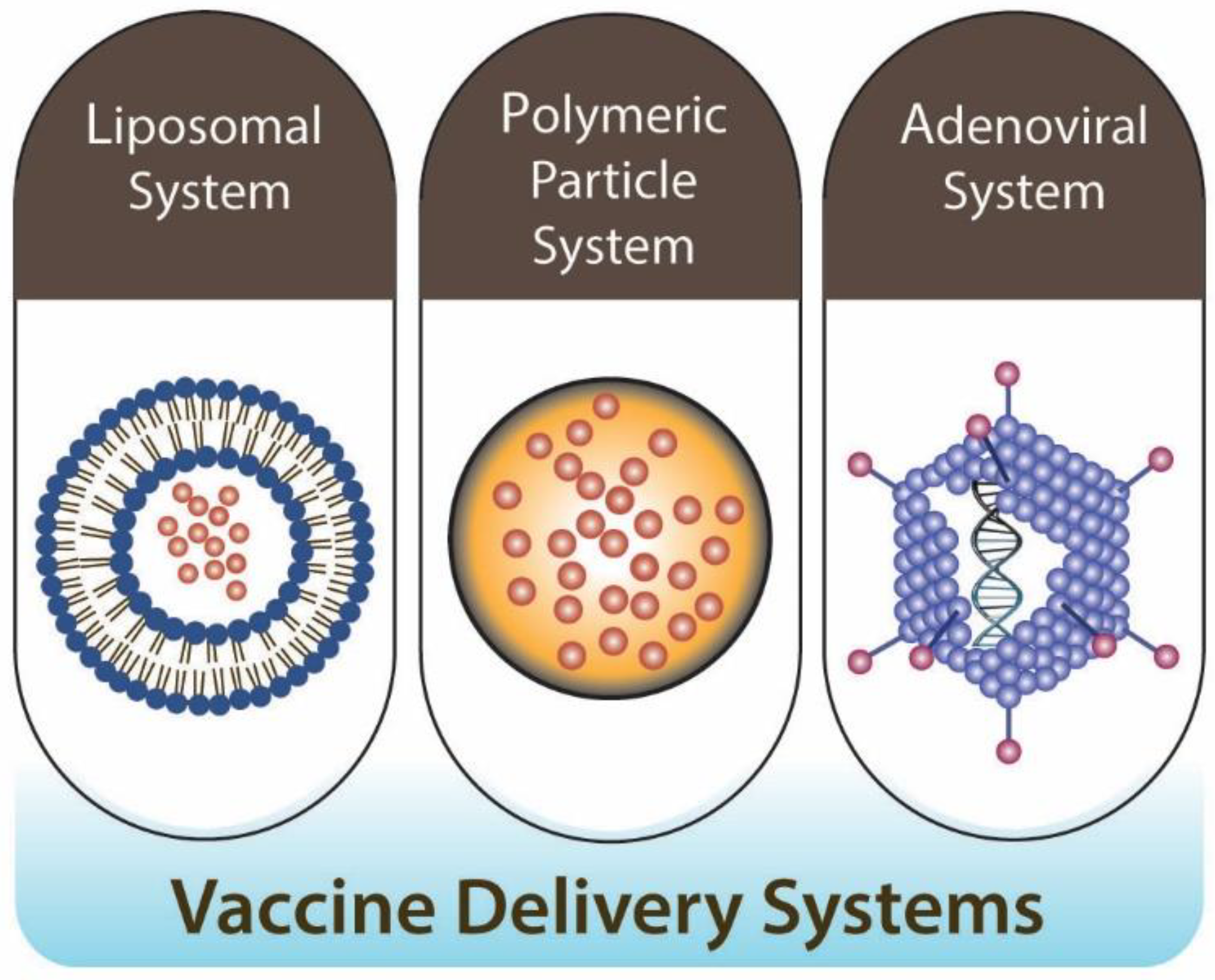
4. Current Oral Vaccine Delivery Systems
4.1. Delivery Strategies of Oral Vaccines
4.2. Liposomal System
4.3. Polymeric Particle System
4.4. Adenoviral System
5. Direction to Enhance Oral Vaccination
5.1. Biological Target-M Cell
5.2. Self-Adjuvants
5.3. Delivery System-Hybrid Delivery System
6. Conclusions and Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- Janeway, C.A.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Azizi, A.; Kumar, A.; Diaz-Mitoma, F.; Mestecky, J. Enhancing oral vaccine potency by targeting intestinal m cells. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Attarwala, H.; Han, M.; Kim, J.; Amiji, M. Oral nucleic acid therapy using multicompartmental delivery systems. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Fooks, A.R. Development of oral vaccines for human use. Curr. Opin. Mol. Ther. 2000, 2, 80–86. [Google Scholar] [PubMed]
- Marasini, N.; Skwarczynski, M.; Toth, I. Oral delivery of nanoparticle-based vaccines. Expert Rev. Vaccines 2014, 13, 1361–1376. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, H.; Kett, V. Current prospects and future challenges for nasal vaccine delivery. Hum. Vaccines Immunother. 2017, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, J.; Czerkinsky, C. Mucosal immunity and vaccines. Nat. Med. 2005, 11, S45–S53. [Google Scholar] [CrossRef] [PubMed]
- Mestecky, J.; Moldoveanu, Z.; Russell, M.W. Immunologic uniqueness of the genital tract: Challenge for vaccine development. Am. J. Reprod. Immunol. 2005, 53, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, P.A.; Cu-Uvin, S.; Neutra, M.R.; Flanigan, T.P. Comparison of the oral, rectal, and vaginal immunization routes for induction of antibodies in rectal and genital tract secretions of women. Infect. Immun. 1997, 65, 1387–1394. [Google Scholar] [PubMed]
- Kulkarni, P.S.; Raut, S.K.; Dhere, R.M. A post-marketing surveillance study of a human live-virus pandemic influenza a (h1n1) vaccine (nasovac (r)) in India. Hum. Vaccines Immunother. 2013, 9, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J.; Curran, M.P. Live attenuated influenza vaccine (flumist(r); fluenz): A review of its use in the prevention of seasonal influenza in children and adults. Drugs 2011, 71, 1591–1622. [Google Scholar] [CrossRef] [PubMed]
- Miyake, A.; Akagi, T.; Enose, Y.; Ueno, M.; Kawamura, M.; Horiuchi, R.; Hiraishi, K.; Adachi, M.; Serizawa, T.; Narayan, O.; et al. Induction of hiv-specific antibody response and protection against vaginal shiv transmission by intranasal immunization with inactivated shiv-capturing nanospheres in macaques. J. Med. Virol. 2004, 73, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. Overview of the mucosal immune system. Curr. Top. Microbiol. Immunol. 1989, 146, 13–25. [Google Scholar] [PubMed]
- Garside, P.; Mowat, A.M. Oral tolerance. Semin. Immunol. 2001, 13, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wilkhu, J.; McNeil, S.E.; Kirby, D.J.; Perrie, Y. Formulation design considerations for oral vaccines. Ther. Deliv. 2011, 2, 1141–1164. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, W.S. Development of oral vaccines to stimulate mucosal and systemic immunity: Barriers and novel strategies. Clin. Immunol. Immunopathol. 1995, 74, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Krishnan, L.; Omri, A. Nasal and pulmonary vaccine delivery using particulate carriers. Expert Opin. Drug Deliv. 2015, 12, 993–1008. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Pandey, A.N.; Jain, S.K. Nasal-nanotechnology: Revolution for efficient therapeutics delivery. Drug Deliv. 2016, 23, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Nizard, M.; Diniz, M.O.; Roussel, H.; Tran, T.; Ferreira, L.C.S.; Badoual, C.; Tartour, E. Mucosal vaccines. Hum. Vaccines Immunother. 2014, 10, 2175–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsch, M.; Zhou, W.; Rhodes, P.; Bopp, M.; Chen, R.T.; Linder, T.; Spyr, C.; Steffen, R. Use of the inactivated intranasal influenza vaccine and the risk of bell’s palsy in switzerland. N. Engl. J. Med. 2004, 350, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Watts, P.; Smith, A.; Hinchcliffe, M. Chisys® as a Chitosan-Based Delivery Platform for Nasal Vaccination. In Mucosal Delivery of Biopharmaceuticals: Biology, Challenges and Strategies; das Neves, J., Sarmento, B., Eds.; Springer: Boston, MA, USA, 2014; pp. 499–516. [Google Scholar]
- Shakya, A.K.; Chowdhury, M.Y.E.; Tao, W.; Gill, H.S. Mucosal vaccine delivery: Current state and a pediatric perspective. J. Control. Release 2016, 240, 394–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela Ramirez, J.E.; Sharpe, L.A.; Peppas, N.A. Current state and challenges in developing oral vaccines. Adv. Drug Deliv. Rev. 2017, 114, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.N.; Cravioto, A.; Sur, D.; Kanungo, S. Maximizing protection from use of oral cholera vaccines in developing country settings. Hum. Vaccines Immunother. 2014, 10, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandola, T.R.; Taneja, S.; Goyal, N.; Antony, K.; Bhatia, K.; More, D.; Bhandari, N.; Cho, I.; Mohan, K.; Prasad, S.; et al. Rotavac® does not interfere with the immune response to childhood vaccines in indian infants: A randomized placebo controlled trial. Heliyon 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Kuate Defo, Z.; Lee, B. New approaches in oral rotavirus vaccines. Crit. Rev. Microbiol. 2016, 42, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, J.-Y.; Wang, X.; Chen, J.-T.; Xia, M.; Hu, W.; Zou, Y.; Yin, W.-D. Review of 10 years of clinical experience with chinese domestic trivalent influenza vaccine anflu®. Hum. Vaccines Immunother. 2014, 10, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.N.; Pezzoli, L.; Alberti, K.P.; Martin, S.; Costa, A.; Perea, W.; Legros, D. Achievements and challenges for the use of killed oral cholera vaccines in the global stockpile era. Hum. Vaccines Immunother. 2017, 13, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, A.; Desai, S.N.; Mogasale, V.; Excler, J.-L.; Digilio, L. Lessons learnt from 12 oral cholera vaccine campaigns in resource-poor settings. Bull. World Health Organ. 2017, 95, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Gutiérrez, L.; Soriano, V.; Requena, S.; Arias, A.; Barreiro, P.; de Mendoza, C. Treatment and prevention of hiv infection with long-acting antiretrovirals. Expert Rev. Clin. Pharmacol. 2018, 11, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.A.; Gonzalez-Garcia, J.; Stellbrink, H.-J.; Eron, J.J.; Yazdanpanah, Y.; Podzamczer, D.; Lutz, T.; Angel, J.B.; Richmond, G.J.; Clotet, B.; et al. Long-acting intramuscular cabotegravir and rilpivirine in adults with hiv-1 infection (latte-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet 2017, 390, 1499–1510. [Google Scholar] [CrossRef]
- Ratanasuwan, W.; Kim, Y.H.; Sah, B.K.; Suwanagool, S.; Kim, D.R.; Anekthananon, A.; Lopez, A.L.; Techasathit, W.; Grahek, S.L.; Clemens, J.D.; et al. Peru-15 (choleragarde®), a live attenuated oral cholera vaccine, is safe and immunogenic in human immunodeficiency virus (hiv)-seropositive adults in Thailand. Vaccine 2015, 33, 4820–4826. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J.; Jepson, M.A.; Baird, A.W. Keynote review: Intestinal peyer’s patch m cells and oral vaccine targeting. Drug Discov. Today 2005, 10, 1145–1157. [Google Scholar] [CrossRef]
- Mowat, A.M. Anatomical basis of tolerance and immunity to intestinal antigens. Nat. Rev. Immunol. 2003, 3, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Khoo, U.Y.; Proctor, I.E.; Macpherson, A.J. CD4+ t cell down-regulation in human intestinal mucosa: Evidence for intestinal tolerance to luminal bacterial antigens. J. Immunol. 1997, 158, 3626–3634. [Google Scholar] [PubMed]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory t lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Hussain, N. Regulatory aspects in the pharmaceutical development of nanoparticle drug delivery systems designed to cross the intestinal epithelium and m-cells. Int. J. Pharm. 2016, 514, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Mojaverian, P.; Ferguson, R.K.; Vlasses, P.H.; Rocci, M.L., Jr.; Oren, A.; Fix, J.A.; Caldwell, L.J.; Gardner, C. Estimation of gastric residence time of the Heidelberg capsule in humans: Effect of varying food composition. Gastroenterology 1985, 89, 392–397. [Google Scholar] [CrossRef]
- Gill, N.; Wlodarska, M.; Finlay, B.B. Roadblocks in the gut: Barriers to enteric infection. Cell Microbiol. 2011, 13, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, K.Y.; Jang, Y.S. Mucosal immune system and m cell-targeting strategies for oral mucosal vaccination. Immune Netw 2012, 12, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Wang, L.; Yang, T.; Ma, G.; Wang, S. M-cell targeted polymeric lipid nanoparticles containing a toll-like receptor agonist to boost oral immunity. Int. J. Pharm. 2014, 473, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.A.; Jepson, M.A.; Hirst, B.H. Exploiting m cells for drug and vaccine delivery. Adv. Drug Deliv. Rev. 2001, 50, 81–106. [Google Scholar] [CrossRef]
- Kuolee, R.; Chen, W. M cell-targeted delivery of vaccines and therapeutics. Expert Opin. Drug Deliv. 2008, 5, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Buda, A.; Sands, C.; Jepson, M.A. Use of fluorescence imaging to investigate the structure and function of intestinal m cells. Adv. Drug Deliv. Rev. 2005, 57, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Meynell, H.M.; Thomas, N.W.; James, P.S.; Holland, J.; Taussig, M.J.; Nicoletti, C. Up-regulation of microsphere transport across the follicle-associated epithelium of peyer’s patch by exposure to streptococcus pneumoniae r36a. FASEB J. 1999, 13, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.S.; Garon, J.; Seib, K.; Orenstein, W.A. Polio vaccination: Past, present and future. Future Microbiol. 2015, 10, 791–808. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kuchroo, V.K.; Inobe, J.; Hafler, D.A.; Weiner, H.L. Regulatory t cell clones induced by oral tolerance: Suppression of autoimmune encephalomyelitis. Science 1994, 265, 1237–1240. [Google Scholar] [CrossRef] [PubMed]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+ t-cell subset inhibits antigen-specific t-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Polanski, M.; Melican, N.S.; Zhang, J.; Weiner, H.L. Oral administration of the immunodominant b-chain of insulin reduces diabetes in a co-transfer model of diabetes in the nod mouse and is associated with a switch from th1 to th2 cytokines. J. Autoimmun. 1997, 10, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Thornton, A.M.; Shevach, E.M. CD4+CD25+ immunoregulatory t cells suppress polyclonal t cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998, 188, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated t cells expressing il-2 receptor alpha-chains (cd25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Chen, Y.; Inobe, J.; Marks, R.; Gonnella, P.; Kuchroo, V.K.; Weiner, H.L. Peripheral deletion of antigen-reactive t cells in oral tolerance. Nature 1995, 376, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Whitacre, C.C.; Gienapp, I.E.; Orosz, C.G.; Bitar, D.M. Oral tolerance in experimental autoimmune encephalomyelitis. Iii. Evidence for clonal anergy. J. Immunol. 1991, 147, 2155–2163. [Google Scholar] [PubMed]
- Kim, L.; Martinez, C.J.; Hodgson, K.A.; Trager, G.R.; Brandl, J.R.; Sandefer, E.P.; Doll, W.J.; Liebowitz, D.; Tucker, S.N. Systemic and mucosal immune responses following oral adenoviral delivery of influenza vaccine to the human intestine by radio controlled capsule. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.H.; Lee, S.E.; Kim, S.Y. Mucosal vaccine adjuvants update. Clin. Exp. Vaccine Res. 2012, 1, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Davitt, C.J.H.; Lavelle, E.C. Delivery strategies to enhance oral vaccination against enteric infections. Adv. Drug Deliv. Rev. 2015, 91, 52–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leder, B.Z. Chapter 61-combination osteoporosis therapy with parathyroid hormone. In The Parathyroids, 3rd ed.; Bilezikian, J.P., Ed.; Academic Press: San Diego, CA, USA, 2015; pp. 853–863. [Google Scholar]
- Irvine, D.J.; Hanson, M.C.; Rakhra, K.; Tokatlian, T. Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev. 2015, 115, 11109–11146. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, T.A.S.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.S.; Alonso, M.J.; Brayden, D.J. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Wang, D.; Mei, D.; Zhang, H.; Wang, Z.; He, B.; Dai, W.; Zhang, H.; Wang, X.; Zhang, Q. Macrophage mediated biomimetic delivery system for the treatment of lung metastasis of breast cancer. J. Control. Release 2015, 204, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Wang, L.; Yang, T.; Ma, G.; Wang, S. Homogeneous plga-lipid nanoparticle as a promising oral vaccine delivery system for ovalbumin. Asian J. Pharm. Sci. 2014, 9, 129–136. [Google Scholar] [CrossRef]
- Chadwick, S.; Kriegel, C.; Amiji, M. Nanotechnology solutions for mucosal immunization. Adv. Drug Deliv. Rev. 2010, 62, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Devriendt, B.; De Geest, B.G.; Goddeeris, B.M.; Cox, E. Crossing the barrier: Targeting epithelial receptors for enhanced oral vaccine delivery. J. Control. Release 2012, 160, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Harsløf, T.; Langdahl, B.L. New horizons in osteoporosis therapies. Curr. Opin. Pharm. 2016, 28, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Peek, L.J.; Middaugh, C.R.; Berkland, C. Nanotechnology in vaccine delivery. Adv. Drug Deliv. Rev. 2008, 60, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Harde, H.; Indulkar, A.; Agrawal, A.K. Improved stability and immunological potential of tetanus toxoid containing surface engineered bilosomes following oral administration. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, T.; Zhang, M.; Chen, R.; Niu, R.; Deng, Y. Mannose derivative and lipid a dually decorated cationic liposomes as an effective cold chain free oral mucosal vaccine adjuvant-delivery system. Eur. J. Pharm. Biopharm. 2014, 88, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hong, Y.; Chen, W.; Wang, C. Polymers for DNA vaccine delivery. ACS Biomater. Sci. Eng. 2017, 3, 108–125. [Google Scholar] [CrossRef]
- Duran-Lobato, M.; Carrillo-Conde, B.; Khairandish, Y.; Peppas, N.A. Surface-modified p(hema-co-maa) nanogel carriers for oral vaccine delivery: Design, characterization, and in vitro targeting evaluation. Biomacromolecules 2014, 15, 2725–2734. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Maharjan, S.; Jiang, T.; Kang, S.K.; Choi, Y.J.; Cho, C.S. Combinatorial approach of antigen delivery using m cell-homing peptide and mucoadhesive vehicle to enhance the efficacy of oral vaccine. Mol. Pharm. 2015, 12, 3816–3828. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, L.A.; Vela Ramirez, J.E.; Haddadin, O.M.; Ross, K.A.; Narasimhan, B.; Peppas, N.A. Ph-responsive microencapsulation systems for the oral delivery of polyanhydride nanoparticles. Biomacromolecules 2018, 19, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Li, P.; Deng, J.; Gao, N.; Zhang, Y.; Pan, H.; Liu, L.; Wang, C.; Cai, L.; Ma, Y. Cationic polypeptide micelle-based antigen delivery system: A simple and robust adjuvant to improve vaccine efficacy. J. Control. Release 2013, 170, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Maharjan, S.; Jiang, T.; Kang, S.K.; Choi, Y.J.; Cho, C.S. Attuning hydroxypropyl methylcellulose phthalate to oral delivery vehicle for effective and selective delivery of protein vaccine in ileum. Biomaterials 2015, 59, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Liu, W.; Liu, H.; Li, C.; Zhang, Y.; Meng, X.; Tang, T.; Xi, T.; Xing, Y. Oral helicobacter pylori vaccine-encapsulated acid-resistant hp55/plga nanoparticles promote immune protection. Eur. J. Pharm. Biopharm. 2017, 111, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, C.; Yang, W.; Liu, X.; Wu, Y. Chemical synthesis, versatile structures and functions of tailorable adjuvants for optimizing oral vaccination. ACS Appl. Mater. Interfaces 2016, 8, 34933–34950. [Google Scholar] [CrossRef] [PubMed]
- Dinda, A.K.; Bhat, M.; Srivastava, S.; Kottarath, S.K.; Prashant, C.K. Novel nanocarrier for oral hepatitis b vaccine. Vaccine 2016, 34, 3076–3081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zeng, Z.; Hu, C.; Bellis, S.L.; Yang, W.; Su, Y.; Zhang, X.; Wu, Y. Controlled and targeted release of antigens by intelligent shell for improving applicability of oral vaccines. Biomaterials 2016, 77, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Singh, B.; Li, H.S.; Kim, Y.K.; Kang, S.K.; Nah, J.W.; Choi, Y.J.; Cho, C.S. Targeted oral delivery of bmpb vaccine using porous plga microparticles coated with m cell homing peptide-coupled chitosan. Biomaterials 2014, 35, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Suksamran, T.; Ngawhirunpat, T.; Rojanarata, T.; Sajomsang, W.; Pitaksuteepong, T.; Opanasopit, P. Methylated N-(4-N,N-dimethylaminocinnamyl) chitosan-coated electrospray ova-loaded microparticles for oral vaccination. Int. J. Pharm. 2013, 448, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Baert, K.; de Geest, B.G.; de Rycke, R.; da Fonseca Antunes, A.B.; de Greve, H.; Cox, E.; Devriendt, B. Beta-glucan microparticles targeted to epithelial apn as oral antigen delivery system. J. Control. Release 2015, 220, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Guillén, D.; Moreno-Mendieta, S.; Pérez, R.; Espitia, C.; Sánchez, S.; Rodríguez-Sanoja, R. Starch granules as a vehicle for the oral administration of immobilized antigens. Carbohydr. Polym. 2014, 112, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Modi, A.; Sajjan, S.; Michael Lewiecki, E.; Harris, S.T.; Papadopoulos Weaver, J. Relationship between gastrointestinal events and compliance with osteoporosis therapy: An administrative claims analysis of the us managed care population. Clin. Ther. 2016, 38, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Singh, B.; Maharjan, S.; Li, H.-S.; Kang, S.-K.; Bok, J.-D.; Cho, C.-S.; Choi, Y.-J. Oral delivery of probiotic expressing m cell homing peptide conjugated bmpb vaccine encapsulated into alginate/chitosan/alginate microcapsules. Eur. J. Pharm. Biopharm. 2014, 88, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Barhate, G.; Gautam, M.; Gairola, S.; Jadhav, S.; Pokharkar, V. Enhanced mucosal immune responses against tetanus toxoid using novel delivery system comprised of chitosan-functionalized gold nanoparticles and botanical adjuvant: Characterization, immunogenicity, and stability assessment. J. Pharm. Sci. 2014, 103, 3448–3456. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Yue, Y.; Fan, X.; Dong, C.; Xu, W.; Xiong, S. M cell-targeting strategy facilitates mucosal immune response and enhances protection against cvb3-induced viral myocarditis elicited by chitosan-DNA vaccine. Vaccine 2014, 32, 4457–4465. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Chattopadhyay, M.; Sen, K.K.; Saha, M.K. Development and characterization of alginate coated low molecular weight chitosan nanoparticles as new carriers for oral vaccine delivery in mice. Carbohydr. Polym. 2015, 121, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Cole, H.; Bryan, D.; Lancaster, L.; Mawas, F.; Vllasaliu, D. Chitosan nanoparticle antigen uptake in epithelial monolayers can predict mucosal but not systemic in vivo immune response by oral delivery. Carbohydr. Polym. 2018, 190, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Xia, G.; Bao, Z.; Feng, C.; Cheng, X.; Kong, M.; Liu, Y.; Chen, X. Chitosan based nanoparticles as protein carriers for efficient oral antigen delivery. Int. J. Biol. Macromol. 2016, 91, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, F.-Q.; Shah, Z.; Cheng, X.-J.; Kong, M.; Feng, C.; Chen, X.-G. Nano-polyplex based on oleoyl-carboxymethy-chitosan (ocmcs) and hyaluronic acid for oral gene vaccine delivery. Colloids Surf. B 2016, 145, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Zhang, W.; Chen, Y.; Xu, Y.; Wang, B.; Zong, L. Eudragit(r) l100-coated mannosylated chitosan nanoparticles for oral protein vaccine delivery. Int. J. Biol. Macromol. 2018, 113, 534–542. [Google Scholar] [CrossRef] [PubMed]
- De Smet, R.; Demoor, T.; Verschuere, S.; Dullaers, M.; Ostroff, G.R.; Leclercq, G.; Allais, L.; Pilette, C.; Dierendonck, M.; De Geest, B.G.; et al. Beta-glucan microparticles are good candidates for mucosal antigen delivery in oral vaccination. J. Control. Release 2013, 172, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Okazaki, M.; Ohno, N.; Yadomae, T. Enhancement of cytokine production by macrophages stimulated with (1-->3)-beta-d-glucan, grifolan (grn), isolated from grifola frondosa. Biol. Pharm. Bull. 1994, 17, 1554–1560. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.L. Overview of (1→>3)-beta-d-glucan immunobiology. Mediat. Inflamm. 1997, 6, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Nurunnabi, M.; Kang, S.H.; Nafiujjaman, M.; Huh, K.M.; Lee, Y.K.; Kim, Y.C. Oral gavage delivery of pr8 antigen with beta-glucan-conjugated grgds carrier to enhance m-cell targeting ability and induce immunity. Biomacromolecules 2017, 18, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Takafuji, E.T.; Gaydos, J.C.; Allen, R.G.; Top, J.F.H. Simultaneous administration of live, enteric-coated adenovirus types 4,7, and 21 vaccines: Safety and immunogenicity. J. Infect. Dis. 1979, 140, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Deal, C.; Pekosz, A.; Ketner, G. Prospects for oral replicating adenovirus-vectored vaccines. Vaccine 2013, 31, 3236–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loré, K.; Adams, W.C.; Havenga, M.; Precopio, M.L.; Holterman, L.; Goudsmit, J.; Koup, R.A. Myeloid and plasmacytoid dendritic cells are susceptible to recombinant adenovirus vectors and stimulate polyfunctional memory t cell responses. J. Immunol. 2007, 179, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Patterson, L.J.; Kuate, S.; Daltabuit-Test, M.; Li, Q.; Xiao, P.; McKinnon, K.; DiPasquale, J.; Cristillo, A.; Venzon, D.; Haase, A.; et al. Replicating adenovirus-simian immunodeficiency virus (siv) vectors efficiently prime siv-specific systemic and mucosal immune responses by targeting myeloid dendritic cells and persisting in rectal macrophages, regardless of immunization route. Clin. Vaccine Immunol. 2012, 19, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.A.; Blair, H.; Liang, L.; Brey, R.N.; Brayden, D.; Hirst, B.H. Targeting polymerised liposome vaccine carriers to intestinal m cells. Vaccine 2001, 20, 208–217. [Google Scholar] [CrossRef]
- Savidge, T.C.; Smith, M.W.; James, P.S.; Aldred, P. Salmonella-induced m-cell formation in germ-free mouse peyer’s patch tissue. Am. J. Pathol. 1991, 139, 177–184. [Google Scholar] [PubMed]
- Gebert, A.; Steinmetz, I.; Fassbender, S.; Wendlandt, K.H. Antigen transport into peyer’s patches: Increased uptake by constant numbers of m cells. Am. J. Pathol. 2004, 164, 65–72. [Google Scholar] [CrossRef]
- Maharjan, S.; Singh, B.; Jiang, T.; Yoon, S.Y.; Li, H.S.; Kim, G.; Gu, M.J.; Kim, S.J.; Park, O.J.; Han, S.H.; et al. Systemic administration of rankl overcomes the bottleneck of oral vaccine delivery through microfold cells in ileum. Biomaterials 2016, 84, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Giannasca, P.J.; Giannasca, K.T.; Leichtner, A.M.; Neutra, M.R. Human intestinal m cells display the sialyl lewis a antigen. Infect. Immun. 1999, 67, 946–953. [Google Scholar] [PubMed]
- Clark, M.A.; Jepson, M.A.; Simmons, N.L.; Booth, T.A.; Hirst, B.H. Differential expression of lectin-binding sites defines mouse intestinal m-cells. J. Histochem. Cytochem. 1993, 41, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Giannasca, P.J.; Giannasca, K.T.; Falk, P.; Gordon, J.I.; Neutra, M.R. Regional differences in glycoconjugates of intestinal m cells in mice: Potential targets for mucosal vaccines. Am. J. Physiol. 1994, 267, G1108–G1121. [Google Scholar] [CrossRef] [PubMed]
- Roth-Walter, F.; Bohle, B.; Scholl, I.; Untersmayr, E.; Scheiner, O.; Boltz-Nitulescu, G.; Gabor, F.; Brayden, D.J.; Jensen-Jarolim, E. Targeting antigens to murine and human m-cells with aleuria aurantia lectin-functionalized microparticles. Immunol. Lett. 2005, 100, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.; Clark, M.A.; Jepson, M.A.; Hirst, B.H. Ulex europaeus 1 lectin targets microspheres to mouse peyer’s patch m-cells in vivo. Vaccine 1998, 16, 536–541. [Google Scholar] [CrossRef]
- Nochi, T.; Yuki, Y.; Matsumura, A.; Mejima, M.; Terahara, K.; Kim, D.Y.; Fukuyama, S.; Iwatsuki-Horimoto, K.; Kawaoka, Y.; Kohda, T.; et al. A novel m cell-specific carbohydrate-targeted mucosal vaccine effectively induces antigen-specific immune responses. J. Exp. Med. 2007, 204, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- Shima, H.; Watanabe, T.; Fukuda, S.; Fukuoka, S.; Ohara, O.; Ohno, H. A novel mucosal vaccine targeting peyer’s patch m cells induces protective antigen-specific iga responses. Int. Immunol. 2014, 26, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Rochereau, N.; Drocourt, D.; Perouzel, E.; Pavot, V.; Redelinghuys, P.; Brown, G.D.; Tiraby, G.; Roblin, X.; Verrier, B.; Genin, C.; et al. Dectin-1 is essential for reverse transcytosis of glycosylated siga-antigen complexes by intestinal m cells. PLoS Biol. 2013, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochereau, N.; Pavot, V.; Verrier, B.; Ensinas, A.; Genin, C.; Corthesy, B.; Paul, S. Secretory iga as a vaccine carrier for delivery of hiv antigen to m cells. Eur. J. Immunol. 2015, 45, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Kerneis, S.; Bogdanova, A.; Kraehenbuhl, J.P.; Pringault, E. Conversion by peyer’s patch lymphocytes of human enterocytes into m cells that transport bacteria. Science 1997, 277, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Kerneis, S.; Caliot, E.; Stubbe, H.; Bogdanova, A.; Kraehenbuhl, J.; Pringault, E. Molecular studies of the intestinal mucosal barrier physiopathology using cocultures of epithelial and immune cells: A technical update. Microbes Infect. 2000, 2, 1119–1124. [Google Scholar] [CrossRef]
- Lo, D.; Tynan, W.; Dickerson, J.; Scharf, M.; Cooper, J.; Byrne, D.; Brayden, D.; Higgins, L.; Evans, C.; O’Mahony, D.J. Cell culture modeling of specialized tissue: Identification of genes expressed specifically by follicle-associated epithelium of peyer’s patch by expression profiling of caco-2/raji co-cultures. Int. Immunol. 2004, 16, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Lo, D.D.; Ling, J.; Eckelhoefer, A.H. M cell targeting by a claudin 4 targeting peptide can enhance mucosal iga responses. BMC Biotechnol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Rossenu, S.; Dewitte, D.; Vandekerckhove, J.; Ampe, C. A phage display technique for a fast, sensitive, and systematic investigation of protein-protein interactions. J. Protein Chem. 1997, 16, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Fievez, V.; Plapied, L.; Plaideau, C.; Legendre, D.; des Rieux, A.; Pourcelle, V.; Freichels, H.; Jerome, C.; Marchand, J.; Preat, V.; et al. In vitro identification of targeting ligands of human m cells by phage display. Int. J. Pharm. 2010, 394, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.K.; Kang, S.K.; Choi, J.H.; Park, I.K.; Na, H.S.; Lee, H.C.; Kim, E.B.; Lee, N.K.; Nah, J.W.; Choi, Y.J.; et al. Targeted delivery of chitosan nanoparticles to peyer’s patch using m cell-homing peptide selected by phage display technique. Biomaterials 2010, 31, 7738–7747. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, E.C.; O’Hagan, D.T. Delivery systems and adjuvants for oral vaccines. Expert Opin. Drug Deliv. 2006, 3, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Hudalla, G.A.; Modica, J.A.; Tian, Y.F.; Rudra, J.S.; Chong, A.S.; Sun, T.; Mrksich, M.; Collier, J.H. A self-adjuvanting supramolecular vaccine carrying a folded protein antigen. Adv. Healthc. Mater. 2013, 2, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Rudra, J.S.; Tian, Y.F.; Jung, J.P.; Collier, J.H. A self-assembling peptide acting as an immune adjuvant. Proc. Natl. Acad. Sci. USA 2010, 107, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Snook, J.D.; Chesson, C.B.; Peniche, A.G.; Dann, S.M.; Paulucci, A.; Pinchuk, I.V.; Rudra, J.S. Peptide nanofiber-CaCo3 composite microparticles as adjuvant-free oral vaccine delivery vehicles. J. Mater. Chem. B 2016, 4, 1640–1649. [Google Scholar] [CrossRef]
- Zaman, M.; Abdel-Aal, A.B.; Fujita, Y.; Ziora, Z.M.; Batzloff, M.R.; Good, M.F.; Toth, I. Structure-activity relationship for the development of a self-adjuvanting mucosally active lipopeptide vaccine against streptococcus pyogenes. J. Med. Chem. 2012, 55, 8515–8523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, S.; Du, X.; Li, T.; Han, L.; Kong, J. Heterologous expression of carcinoembryonic antigen in lactococcus lactis via lcsb-mediated surface displaying system for oral vaccine development. J. Microbiol. Immunol. Infect. 2016, 49, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Grangette, C.; Muller-Alouf, H.; Geoffroy, M.; Goudercourt, D.; Turneer, M.; Mercenier, A. Protection against tetanus toxin after intragastric administration of two recombinant lactic acid bacteria: Impact of strain viability and in vivo persistence. Vaccine 2002, 20, 3304–3309. [Google Scholar] [CrossRef]
- Jing, H.; Yong, L.; Haiyan, L.; Yanjun, M.; Yun, X.; Yu, Z.; Taiming, L.; Rongyue, C.; Liang, J.; Jie, W.; et al. Oral administration of lactococcus lactis delivered heat shock protein 65 attenuates atherosclerosis in low-density lipoprotein receptor-deficient mice. Vaccine 2011, 29, 4102–4109. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Xu, Y.; Chen, J.; Wei, X.; Lam, D.M. Immunoprotection against influenza h5n1 virus by oral administration of enteric-coated recombinant lactococcus lactis mini-capsules. Virology 2010, 407, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S.; Piao, D.C.; Jiang, T.; Bok, J.D.; Cho, C.S.; Lee, Y.S.; Kang, S.K.; Choi, Y.J. Recombinant interleukin 6 with m cell-targeting moiety produced in lactococcus lactis il1403 as a potent mucosal adjuvant for peroral immunization. Vaccine 2015, 33, 1959–1967. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode of treatment | Name | Desease | Company | Clinical phase | Ref. | |
|---|---|---|---|---|---|---|
| Nasal Vaccines | Current treatment (including Licenses) | FluMist® Quadrivalent | Influenza A subtype and type B viruses | MedImmune, Gaithersburg, MD, USA | Out in Market | [11,17,18,19] |
| Nasovac-STM | Influenza A (H1N1) | Cipla, Mumbai, India | [10,17] | |||
| Nasalflu | Influenza A (H1N1 and H3N2) and type B | Crucell, Leiden, The Netherlands | [20] | |||
| Pre-clinical or clinical trial | μcoTM | Anti-emetic migraine, flu | SNBL, Tokyo, Japan | Phase II, Phase I, pre-clinical | [6] | |
| Optinose | Chronic Rhinosinusitis (CRS), Chronic Sinusitis (CS) | OptiNose, Yardley, PA, USA | Clinical trials (various) | [6] | ||
| Flumis Fleuenz | Flu | MedImmune, Gaithersburg, MD, USA | FDA & EMA | [6] | ||
| ChiSys | Avian influenza virus (H5 and H7 subtypes) | Archimedes Pharma, Reading, UK | Phase I, pre-clinical | [6,21] | ||
| Oral Vaccines | Current treatment (including Licenses) | Dukoral® | Vibrio Cholera | VALNEVA, Lyon, France | Out in Market | [22,23,24] |
| BiopolioTM B1/3 | Types 1, 2 and 3 attenuated poliomyelitis viruses (Sabin Strains) | Bharat Biotech, Telagana, India | [22,25] | |||
| Rotarix® | Rotavirus | GSK, Brentford, UK | [19,22,26] | |||
| RotaTeq® | Rotavirus | MSD, Kenilworth, NJ, USA | [19,22,26] | |||
| Vivotif® | Salmonella typhi | PaxVax, Redwood city, CA, USA | [19,22,23] | |||
| Anflu® | 3 influenza virus strains (H1N1 influenza A virus subtype, H3N2 influenza A virus subtype, influenza B) | Alco Pharma, Dhaka, Bangladesh | [27] | |||
| Euvichol® | Vibrio Cholera | Eubiologics, Seoul, South Korea | [28] | |||
| Cholvax® | Vibrio Cholera | Incepta, Dhaka, Bangladesh | [28,29] | |||
| Shanchol® | Vibrio Cholerae | SANOFI, Paris, Frnace | [19,24,29] | |||
| Pre-clinical or clinical trial | LATTE-2 | Hepatitis C virus | GSK, Brentford, UK | Phase IIb | [30,31] | |
| CholeraGarde® | Human immunodeficiency virus | AVANT Immunotherapeutics, Needham, MA, USA | Phase II | [24,32] | ||
| RV3-BB | Rotavirus | GSK, Brentford, UK | Phase II, Phase I | [26] | ||
| ORV 116E | Rotavirus | SAS, Delhi, India | Phase III, Phase II, Phase I | [26] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.H.; Hong, S.J.; Lee, Y.-K.; Cho, S. Oral Vaccine Delivery for Intestinal Immunity—Biological Basis, Barriers, Delivery System, and M Cell Targeting. Polymers 2018, 10, 948. https://doi.org/10.3390/polym10090948
Kang SH, Hong SJ, Lee Y-K, Cho S. Oral Vaccine Delivery for Intestinal Immunity—Biological Basis, Barriers, Delivery System, and M Cell Targeting. Polymers. 2018; 10(9):948. https://doi.org/10.3390/polym10090948
Chicago/Turabian StyleKang, Sung Hun, Seok Jin Hong, Yong-Kyu Lee, and Sungpil Cho. 2018. "Oral Vaccine Delivery for Intestinal Immunity—Biological Basis, Barriers, Delivery System, and M Cell Targeting" Polymers 10, no. 9: 948. https://doi.org/10.3390/polym10090948
APA StyleKang, S. H., Hong, S. J., Lee, Y.-K., & Cho, S. (2018). Oral Vaccine Delivery for Intestinal Immunity—Biological Basis, Barriers, Delivery System, and M Cell Targeting. Polymers, 10(9), 948. https://doi.org/10.3390/polym10090948