1. Introduction
Oral administration remains the most appropriate, useful, and convenient route for the delivery of most pharmaceutical active agents. However, the main problem of many orally administered drugs and drug delivery systems is overcoming several obstacles before reaching their target sites. Nowadays, the research for approaches to improve the oral bioavailability of low permeable and low soluble compounds for oral administration continues. Using nanoparticles (NPs) as drug delivery system has taken place in the literature as one of these strategies [
1]. By applying nanotechnology to medicine, NPs have been created to mimic or alter biological process [
2]. NPs vary in size, although they are considered to be between 10 and 1000 nanometers (nm) and are generally considered to be between 100 and 500 nm in medical applications [
2,
3]. By means of size, surface properties, and manipulation of the material used, NPs can be developed into therapeutic intelligent systems with increased bioavailability [
2]. NP systems offer many advantages, including: improving the stability of hydrophobic drugs, rendering them suitable for oral administration with increased bioavailability; improving biodistribution and pharmacokinetics, resulting in improved efficacy; reducing side effects as a consequence of favored accumulation at target sites and decreasing toxicity by using biocompatible polymers [
4]. In addition, NP systems can deliver the drug to specific tissues and provide controlled release of the drug. So, this targeted and sustained drug delivery decreases the toxicity and increases the patient’s compliance with less frequent dosing [
2].
Poly (lactic-
co-glycolic-acid) (PLGA) is a copolymer widely used as a matrix for NPs due to its biocompatible and biodegradable characteristics. PLGA is obtained by different ratios of lactic acid and glycolic acid during polymerization resulting in different molecular weights (M
w) and different physical, chemical, and physicochemical properties [
5]. The mechanical strength of PLGA is particularly affected by its M
w. The M
w of PLGA directly affects NP properties such as particle size, entrapment efficiency, release properties, and bioavailability [
6]. When the studies have examined this, it has been reported that the PLGA NPs prepared with Biopharmaceutical Classification System (BCS) class-II (carvedilol [
7], rifampicin [
8], clotrimazole [
9]), BCS class-III (Doxorubicin [
10], Lamivudine [
11]) and BCS Class-IV (docetaxel [
12], paclitaxel [
13]) drugs have increased bioavailability and also extended release.
Chitosan (CS) has been widely used in pharmaceutical and medical areas because of its favorable biological properties such as safety, biocompatibility, biodegradability, low-toxicity, bacteriostatic, fungistatic, hemostatic, anticholesterolemic, and anticancerogenic properties [
14]. CS’s mucoadhesive property, when positively charged, allows the interaction with negatively charged membranes and mucosa, promoting a greater interaction, adhesion, and retention of the pharmaceutical form containing CS close to the intestinal epithelium. Also, CS has the ability to temporarily open the tight junctions of the intestinal epithelium, thereby increasing the drug permeability [
15].
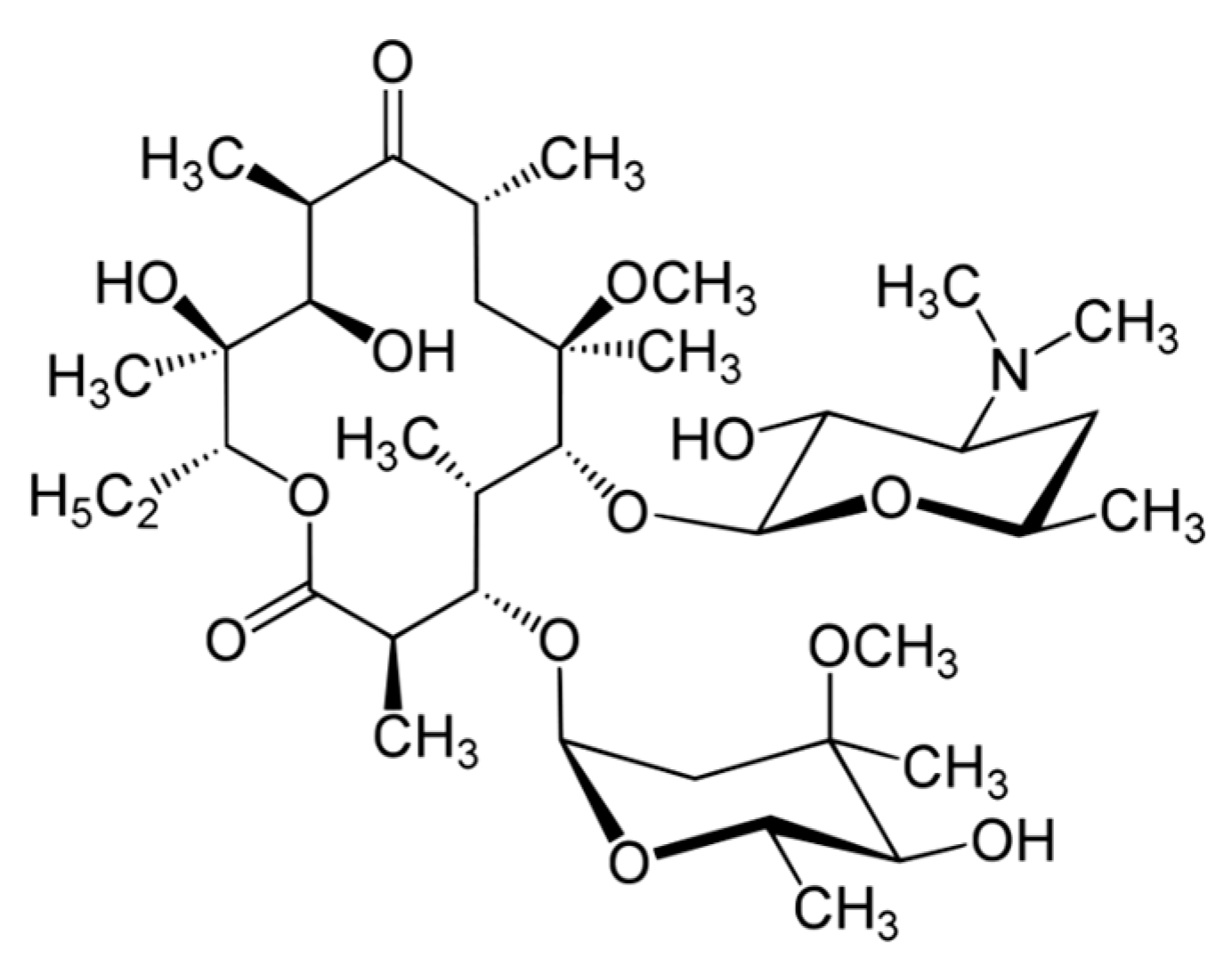
Clarithromycin (CLR) is a semi-synthetic macrolide antibiotic used in many infectious conditions like upper and lower respiratory tracts infections, skin, ear, and other soft tissues infections caused by different bacterial groups. CLR’s chemical formula is C
38H
69NO
13 and the chemical structure of CLR is presented in
Figure 1. CLR is acid stable and has a short half-life (3–4 h) compatible with a twice-a-day administration [
16,
17]. CLR has low systemic oral bioavailability and is a drug belonging to BCS class II. Therefore, short half-life and poor systemic bioavailability of CLR limit the therapeutic efficacy of CLR in intracellular infections. So, a higher CLR dose applied for a longer time to achieve a therapeutic effect that may lead to side and toxic effects such as hepatotoxicity [
16,
17].
So far, CLR-loaded PLGA NPs have been the subject of research in some studies. In one of these studies, NPs were prepared with three different drug: polymer ratios using only Resomer
® RG 502 (lactic acid: glycolic acid ratio 50:50, average M
w: 12.000) by nanoprecipitation technique [
18]. The same study was then supported by intestinal permeability tests [
1]. In another study, CLR-loaded PLGA microspheres were prepared using only one type PLGA (lactic acid: glycolic acid ratio 75:25, M
w 15.000–30.000) by modified O/W single emulsion-solvent evaporation technique [
19]. In another study, CLR-loaded PLGA NPs were prepared by solvent evaporation technique with only one type of PLGA (lactic acid: glycolic acid ratio 50:50) and different drug: polymer ratios [
20]. As can be seen from the previous studies, no studies have examined the effect of M
w of PLGA polymer on CLR-loaded NPs. [
10]. The lactide:glycolide ratio of the PLGA types used in our study was 50:50. The PLGA polymer types were all different in their physical and appearance properties and the lactide:glycolide ratio can completely change the properties expected from the formulation. It is also the fact that the device and surfactant used in preparing the formulation changes the properties of the prepared formulation [
11]. In addition, as can be seen from previous studies, no method used in this study is similar to others and in our study chitosan modification was also made to the formulations.
In this study, we aimed to prepare CLR-loaded PLGA NPs with enhanced bioavailability and increased antibacterial effect with low dose use. For this purpose, we examined three different Mw of PLGA and CS coatings. We prepared three different PLGA NPs and three different CS-coated PLGA NPs by the ‘nanoprecipitation’ technique. The effects of the Mw of PLGA and CS coating on the properties of NPs such as particle size, zeta potential, entrapment efficiency and release rates are discussed in detail. Once these effects were determined, the release kinetics of both burst effect and total release time were examined based on the release results. The crystalline properties of all NPs were then examined by thermal (DSC), FT-IR, and 1H-NMR analysis and finally, antibacterial activity against Staphylococcus aureus (ATCC 25923), Enterococcus faecalis (ATCC 29212), Listeria monocytogenes (ATCC 1911), and Klebsiella pneumoniae (ATCC 700603) was determined by microdilution method. Within the scope of the study, using different Mw of PLGA and coating the formulations with CS is an innovative approach for CLR-loaded PLGA NPs and will add new information to the scientific world.
2. Materials and Methods
2.1. Materials
Clarithromycin was a kind gift from Sanovel (Istanbul, Turkey). Resomer® RG 502 H [Poly (d,l-lactide-co-glycolide), acid-terminated, lactide:glycolide 50:50, Mw: 7.000–17.000], Resomer® RG 503 H [Poly (d,l-lactide-co-glycolide), acid-terminated, lactide:glycolide 50:50, Mw: 24.000–38.000], Resomer® RG 504 H [Poly(d,l-lactide-co-glycolide), acid-terminated, lactide:glycolide 50:50, Mw: 38.000–54.000], and Span® 60 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Low Mw chitosan [Deacetylated chitin/Poly (d-glucosamine), Mw: 50.000–190.000 Da, viscosity: 20–300 cP] was purchased from Sigma (Steinheim, Germany). Pluronic® F-68 was purchased from Alfa-Aesar (Kandel, Germany). All other chemicals used were of analytical grade.
2.2. Preparation of PLGA Nanoparticles and Surface Modification with Chitosan
PLGA-based NPs were prepared by following the nanoprecipitation technique with some modifications [
7]. Briefly, a weighed amount of PLGA (90 mg) was dissolved in 3 mL acetone together with Span
® 60 (30 mg). Next, 3 mL of this solution was added dropwise at a rate of 5 mL.h
−1 into 10 mL of Pluronic
® F-68 aqueous solution (0.5%,
w/
v) under magnetic stirring. Acetone was then allowed to evaporate at room temperature under magnetic stirring for 4 h. The resulting aqueous dispersion was centrifuged to collect the NPs (11.000 rpm, 45 min, 4 °C) (Hettich Rotina-420R, Tuttlingen, Germany). After the NPs were collected, 5 mL of distilled water was added in order to wash the particles. The NPs dispersed in water were again subjected to the above-mentioned centrifugation process. This process was repeated twice to wash the NPs.
For CLR-loaded PLGA-based NP preparation, briefly, the procedure started by adding 9 mg CLR to organic phase. Then, 3 mL of such solution with drugs were added drop-wise at rate 5 mL.h−1 into 10 mL of Pluronic® F-68 aqueous solution (0.5%, w/v) under magnetic stirring. Acetone was then allowed to evaporate at room temperature under magnetic stirring for 4 h. The resulting aqueous dispersion was centrifuged to collect the NPs (11.000 rpm, 45 min, 4 °C) (Hettich Rotina-420R, Tuttlingen, Germany). This process was repeated twice to wash the NPs.
The above procedure was applied with minor modifications when preparing CS-coated formulations. In the CS-coated formulations, the aqueous phase consisted of 10 mL of CS solution (0.25%,
w/
v) and Pluronic
® F-68 (0.5%,
w/
v), both prepared in 2% acetic acid (
v/
v) [
15,
21,
22]. All remaining procedures are the same as above and the formulation ingredients are presented in
Table 1.
2.3. Characterization of Nanoparticles
2.3.1. Particle Size, Polydispersity Index, Zeta Potential
The particle size (PS) and polydispersity index (PDI) of NPs were measured using dynamic light scattering technique on the Zetasizer Nano (Zetasizer Nano ZS, Malvern Instruments, Malvern, UK). PS and PDI of NPs prepared were measured by dispersing the formulation in distilled water. Zeta potential (ZP) was determined using the same instrument in a disposable folded capillary zeta cell at 25 °C room temperature and diluted with distilled water. For statistical analysis all samples were measured in triplicate and the average values and standard deviation of the measurements were calculated.
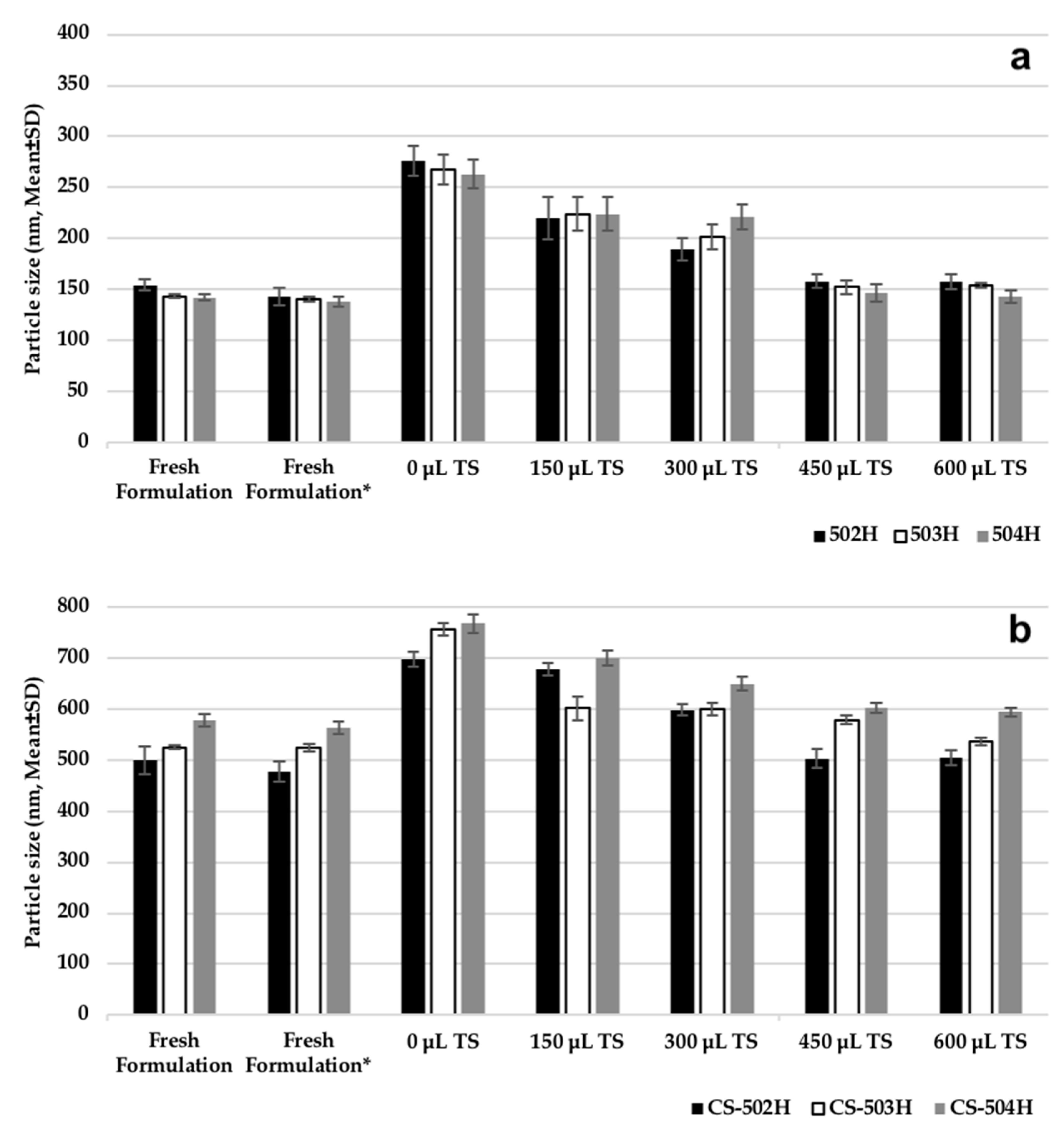
2.3.2. Assessment of Cryoprotectant Effect on Nanoparticles
Some experiments were carried out on PLGA NPs and CS-coated PLGA NP formulations to determine storage and lyophilization conditions. Following PS measurement of fresh formulations, they were centrifuged and the supernatants were discarded. The resulting particles were added to 1 mL of 5% (
w/
v) trehalose solution and the PS measurement was performed again. The dispersion was then divided into five equal portions (200 μL NP suspension) in 5 Eppendorf tubes. While no trehalose solution was added to tube 1, 150 μL, 300 μL, 450 μL, and 600 μL trehalose solution (5%
w/
v) were added to other tubes, respectively. All formulations were then frozen at −20 °C and then lyophilization (Scanvac CoolSafePro Labogene, Lillerød, Denmark) was performed for all tubes, the dry particles were removed from the machine and dispersed in 1 mL of water, followed by PS analysis [
5,
7].
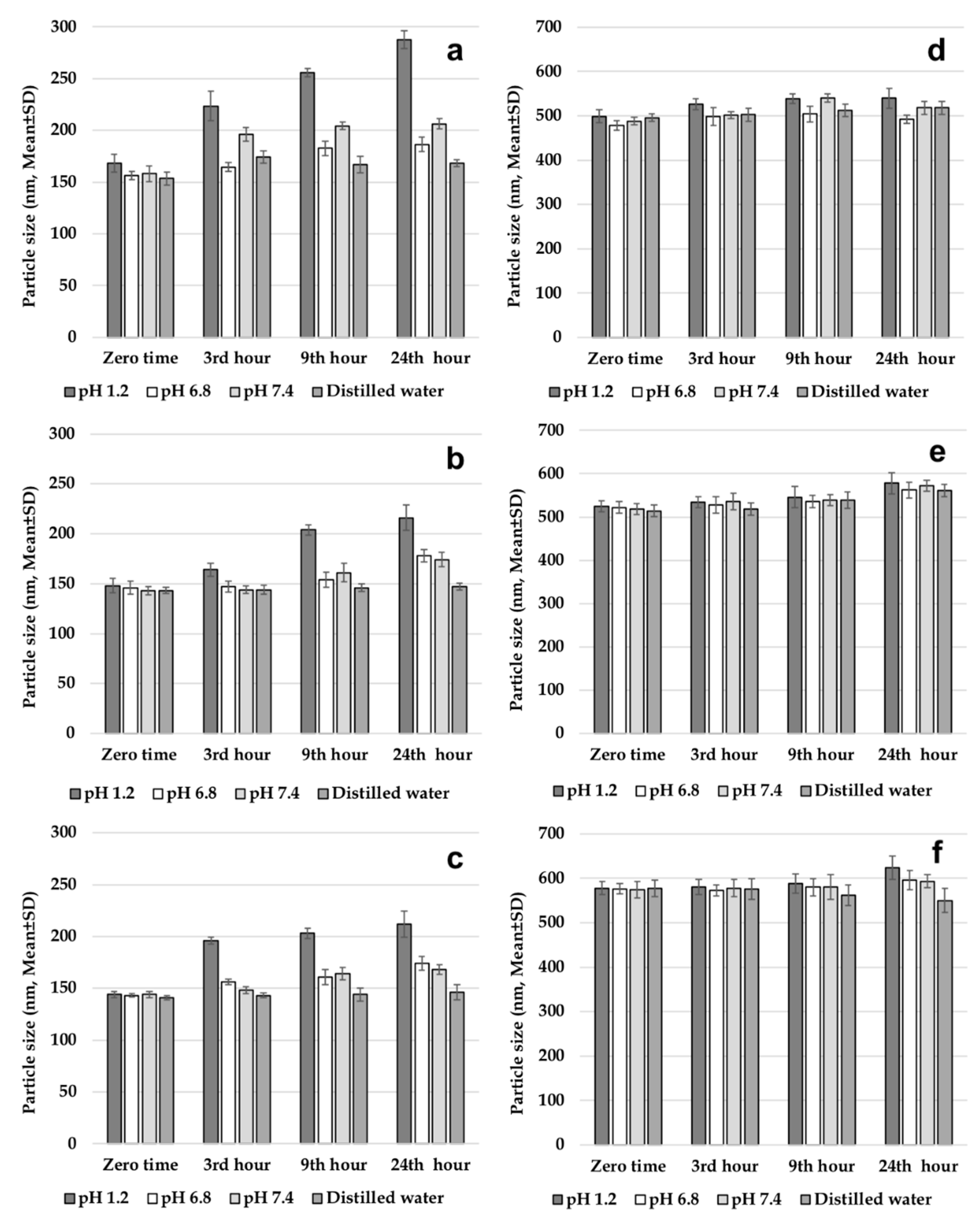
2.3.3. Evaluation of Gastrointestinal Stability of Nanoparticles
Before testing the gastrointestinal stability of NPs, solutions simulating gastrointestinal fluids were prepared. The solutions were pH 1.2 solution; intestinal fluid phosphate buffer solution (pH 6.8); phosphate buffer solution (pH 7.4); and distilled water. All three solutions and distilled water were placed in a shaking water bath at a stirring speed of 40 rpm at a temperature of 37 °C to simulate the gastric medium. One set of formulations (502H, 503H, 504H, CS-502H, CS-503H, CS-504H) was prepared and dispersed in trehalose solution at 5% (
w/
v) concentration. Then, 1 mL of this dispersion was added to solutions incubated at 37 ± 1 °C. Samples were collected after preincubation periods of 3, 9, and 24 h and centrifuged at 4.000 rpm for 5 min to precipitate NPs. Finally, the average PS of the NPs was determined [
5,
7]. This work was performed on fresh nanoparticle batches.
2.3.4. High Performance Liquid Chromatography (HPLC) Conditions
The amount of CLR loaded into NPs and dissolution study of each formulation was performed using HPLC (Shimadzu Corporation, Kyoto, Japan) with reversed-phase Inert Sustain C
18 (5.0 μm, 150 mm × 4.6 mm, GL Sciences Inc., Torrance, CA, USA) column. In the HPLC system, the mobile phase was acetonitrile: 0.035 M potassium dihydrogen phosphate buffer (KH
2PO
4) (55:45
v/
v) mixture, the flow rate was 1 mL.min
−1, while detection was performed at 200 nm at 30 °C. Injection volume was 25 μL. Mobile phase was prepared daily, degassed by sonication, and filtered through 0.45 μm membrane filter before the experiment. The method was validated for precision, accuracy, specificity, and linearity [
23].
2.3.5. Entrapment Efficiency
For entrapment efficiency, the method of extraction of CLR from NPs was used [
5]. Accurately weighed (5 mg) NPs were dissolved in ethyl acetate. The resultant samples were filtered through 0.45 μm membrane filters and analyzed using HPLC. The entrapment efficiency (EE%) of NPs was calculated by Equation (1) [
7].
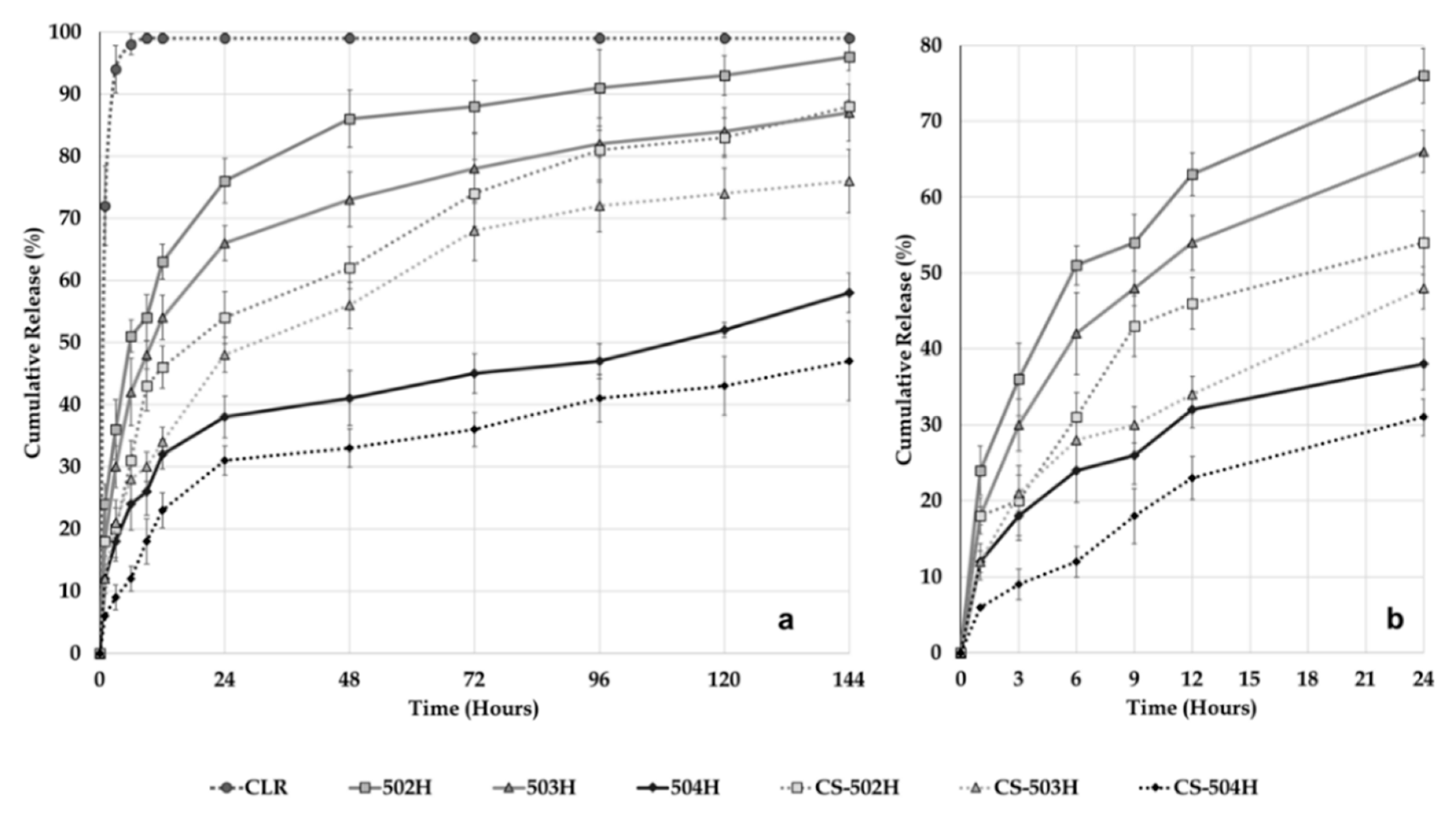
2.3.6. Dissolution and Release Kinetic Evaluation
In vitro dissolution study was performed in intestinal fluid phosphate buffer pH 6.8 containing 1% Tween
® 80 to retain sink conditions maintained at 37 ± 1 °C and 50 rpm using a USP Type II dissolution apparatus (PTWS 820D Pharma Test USP/EP Dissolution Testing Instrument, Hainburg, Germany). Pure CLR (5 mg) and equivalent NP formulations (equivalent 5 mg CLR) were placed in a cellulose acetate dialysis bag (dialysis tubing cellulose membrane, molecular weight cut-off: 14.000, Sigma-Aldrich, USA). After the addition of 1 mL of dissolution medium, the bag was sealed at both ends with special clamps for dissolution. After all the dialysis bags were closed, they were all immersed in the 500 mL of dissolution medium at the same time. Samples of the medium (5 mL) were withdrawn and replaced with fresh medium at 1, 3, 6, 9, 12, 24, 48, 72, 96, 120, and 144 h [
24,
25]. CLR concentration in the samples was analyzed by HPLC. The dissolution study was repeated three times for all NP formulations and pure CLR and the results were calculated as mean ± SD. The results were then plotted as cumulative release.
Data obtained in the
in vitro dissolution studies were further investigated for release kinetics using DDSolver software program. DDSolver computer program was used to shorten the calculation time, eliminate calculation errors, and determine the correct release profile [
26]. DDSolver software program was used for evaluating Higuchi, Korsmeyer-Peppas, Baker-Lonsdale, Peppas-Sahlin, and Weibull models.
2.4. Solid State Characterization of Nanoparticles
2.4.1. Thermal (DSC) Analysis
The physical states of NPs were characterized by differential scanning calorimetry (DSC) (DSC-60, Shimadzu Scientific Instruments, Columbia, MI, USA). Aluminum crucibles with 3 mg samples were analyzed under nitrogen gas (50 mL.min−1) and heating rate of 10 °C.min−1 at a temperature range of 30 and 300 °C. Pure CLR and blank formulation were also analyzed and were used as references.
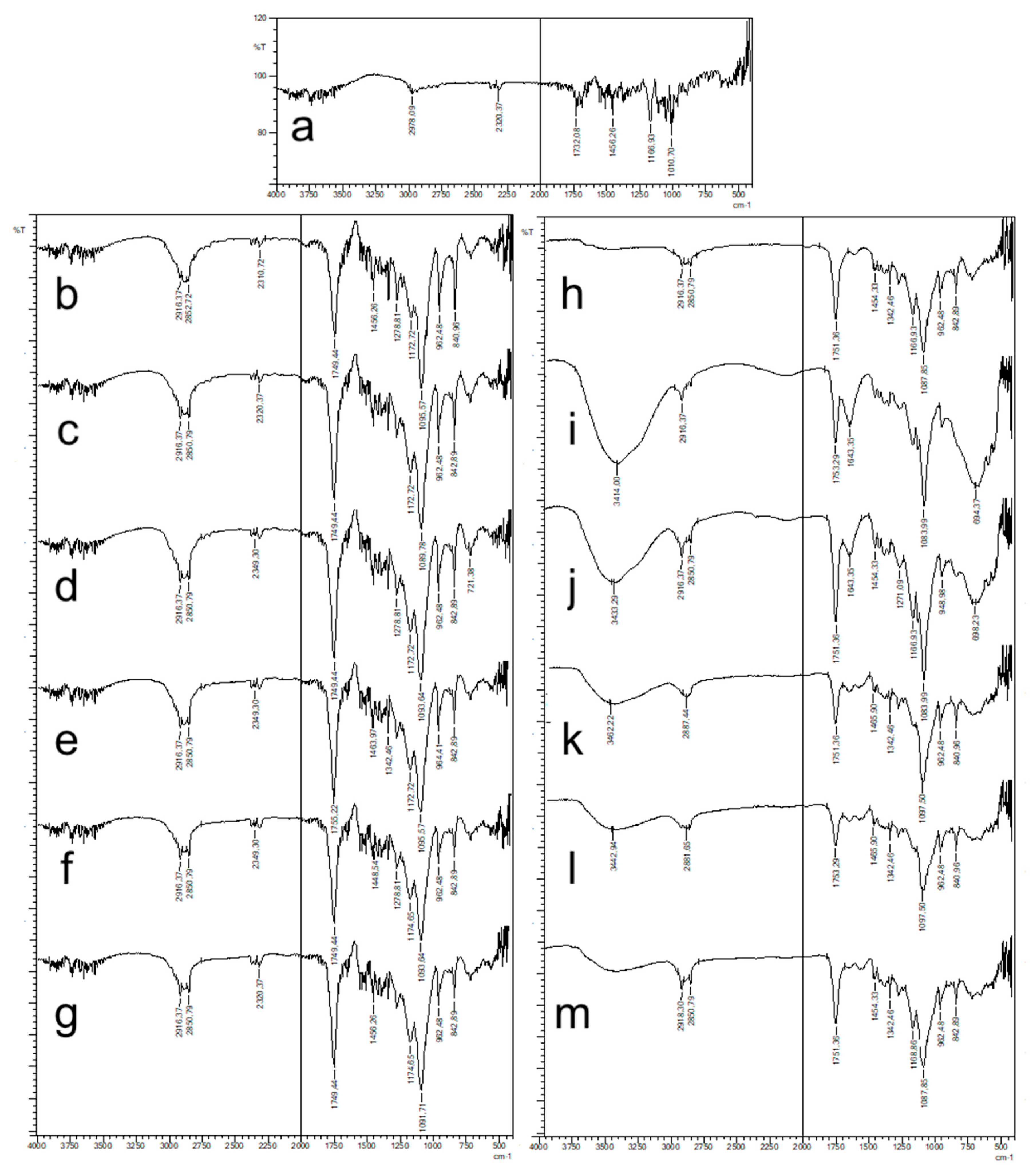
2.4.2. FT-IR Analysis
FT-IR spectra of NPs were recorded using Shimadzu IR Prestige-21 (Shimadzu Corporation, Kyoto, Japan) at the wavelength range of 4000–500 cm−1. Pure CLR and blank formulations were also analyzed and were used as references.
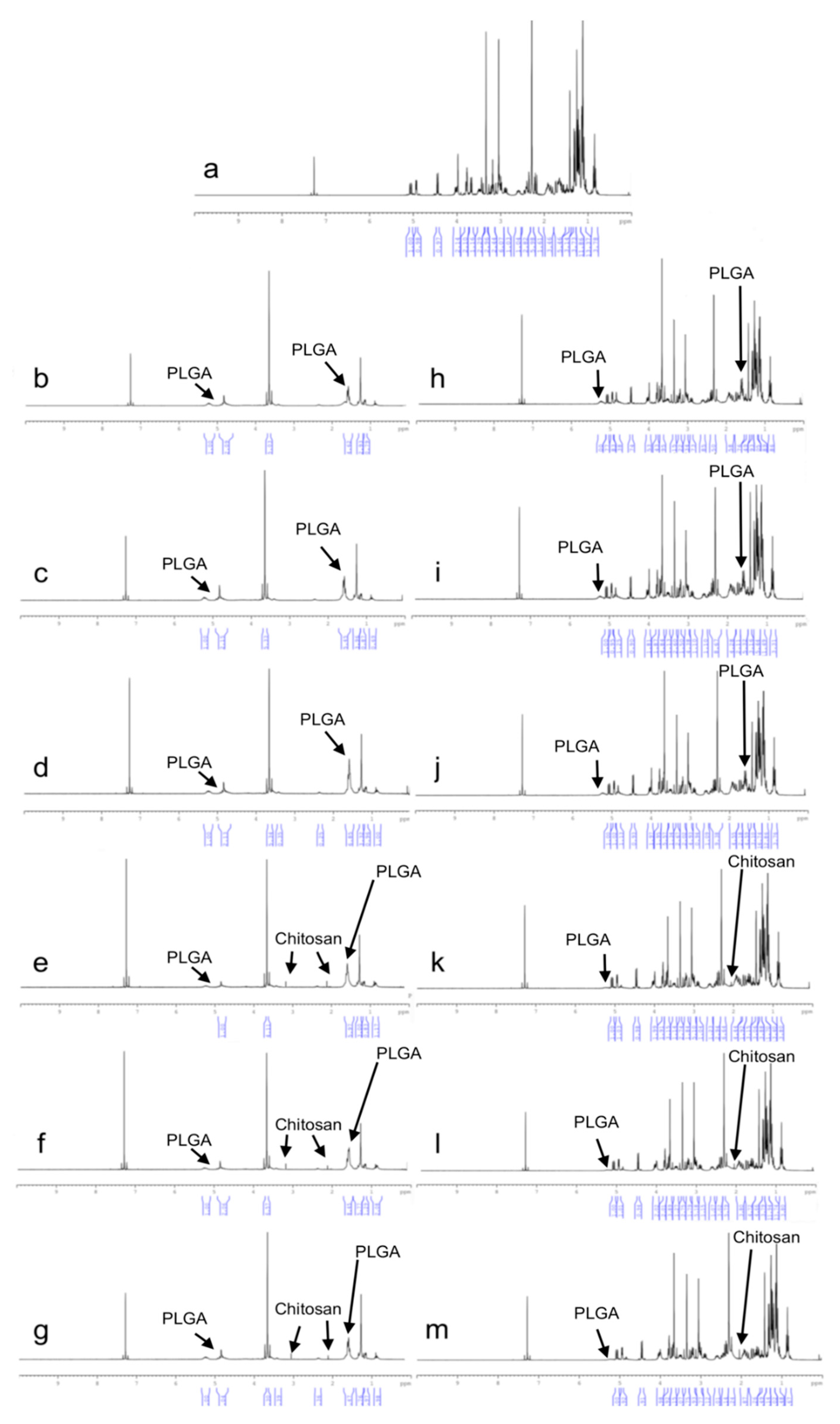
2.4.3. 1H-NMR Analysis
1H-NMR analyses were performed using UltraShieldTM CPMAS NMR (Brucker, Rheinstetten, Germany). Samples were prepared by dissolving formulations in deuterated chloroform (CDCI3). Pure CLR and blank formulations were also analyzed and were used as references.
2.5. Antimicrobial Activity Test
2.5.1. Microorganisms
The following organisms were used in this study: Staphylococcus aureus (ATCC 25923), Enterococcus faecalis (ATCC 29212), Listeria monocytogenes (ATCC 1911), Klebsiella pneumoniae (ATCC 700603). All the bacterial strains were obtained from the ATCC (Rockville, MD, USA). The bacteria were in liquid nutrient broth (Merck) for fresh pure cultures.
2.5.2. Inoculum
The standardization of the bacterial cell number used for susceptibility testing is of critical importance for obtaining accurate and reproducible results. The recommended final inoculum size for broth dilution is 5 × 105 colony-forming units (CFU) per mL. For that reason, all inocula were set in 0.5 McFarland standard with McFarland Tube Densitometer for accurate and reproducible results.
2.5.3. Broth Microdilution Method
Derivatives of substances were dissolved/suspended in DMSO and concentrations were prepared on ranging from 0.49 to 250 µL.mL−1. The prepared concentrations were distributed in duplicate 100 µl for each well on the 96-well plate. After that, fresh pure bacterial cultures, which were set in 0.5 McFarland standard in Mueller Hinton Broth (Sigma-Aldrich), were added at concentrations of 100 µL. After adding the bacterial cultures, final concentrations for all derivatives ranged from 250 to 0.49 µL.mL−1. At the end of this process, all plates were incubated for 24 h. All steps were performed as recommended by CLSI protocol.
2.6. Software Program
Microsoft Excel and DDSolver were employed for calculations.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}