Characteristics of Self-Healable Copolymers of Styrene and Eugenol Terminated Polyurethane Prepolymer




Abstract
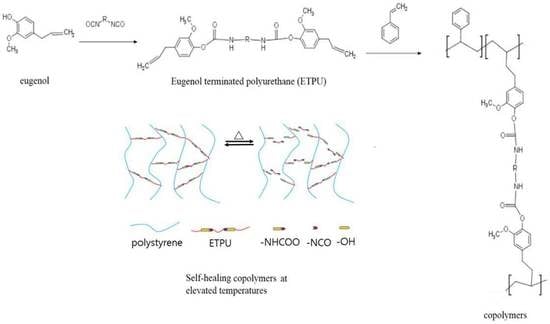
:
1. Introduction
2. Experimentals
2.1. Materials
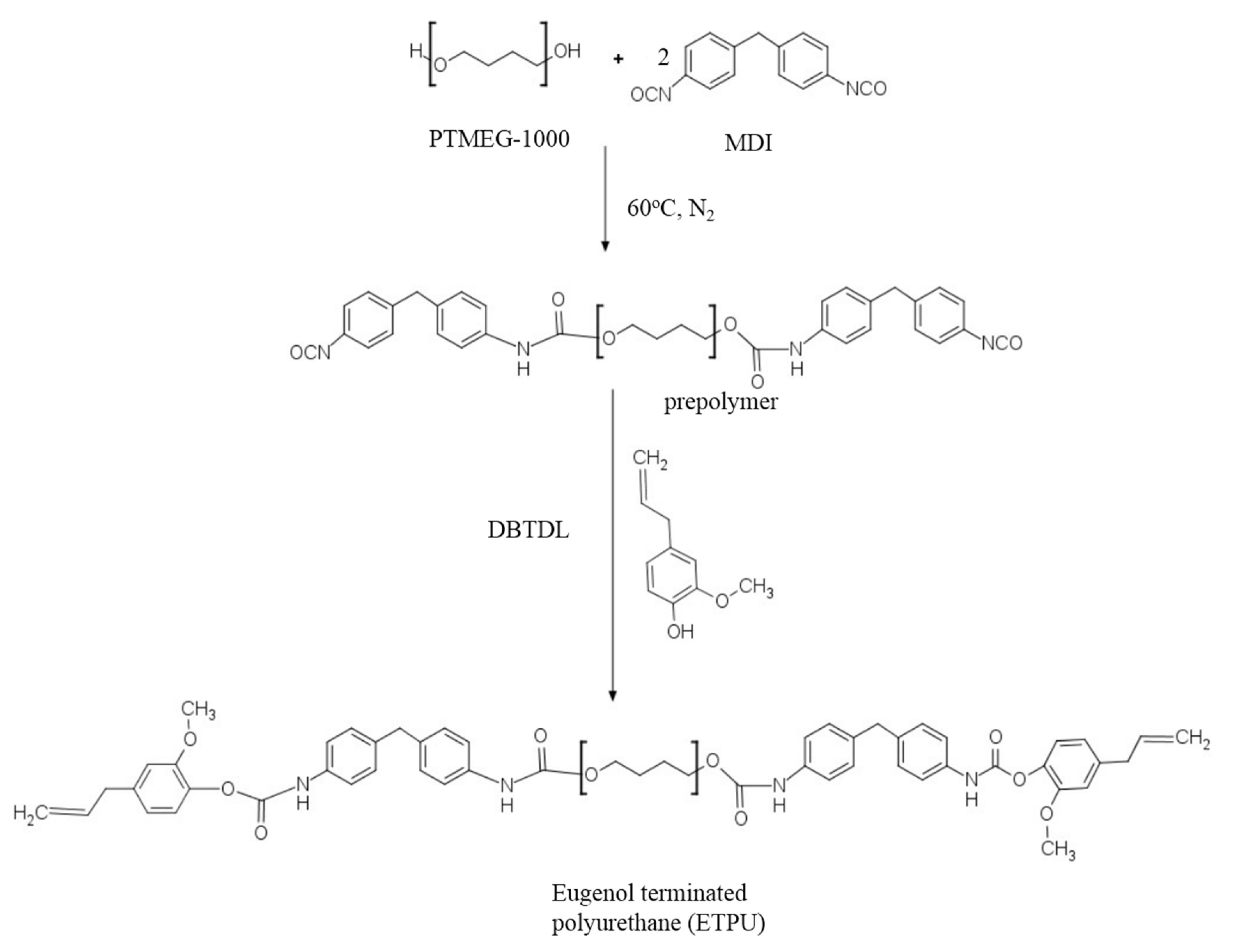
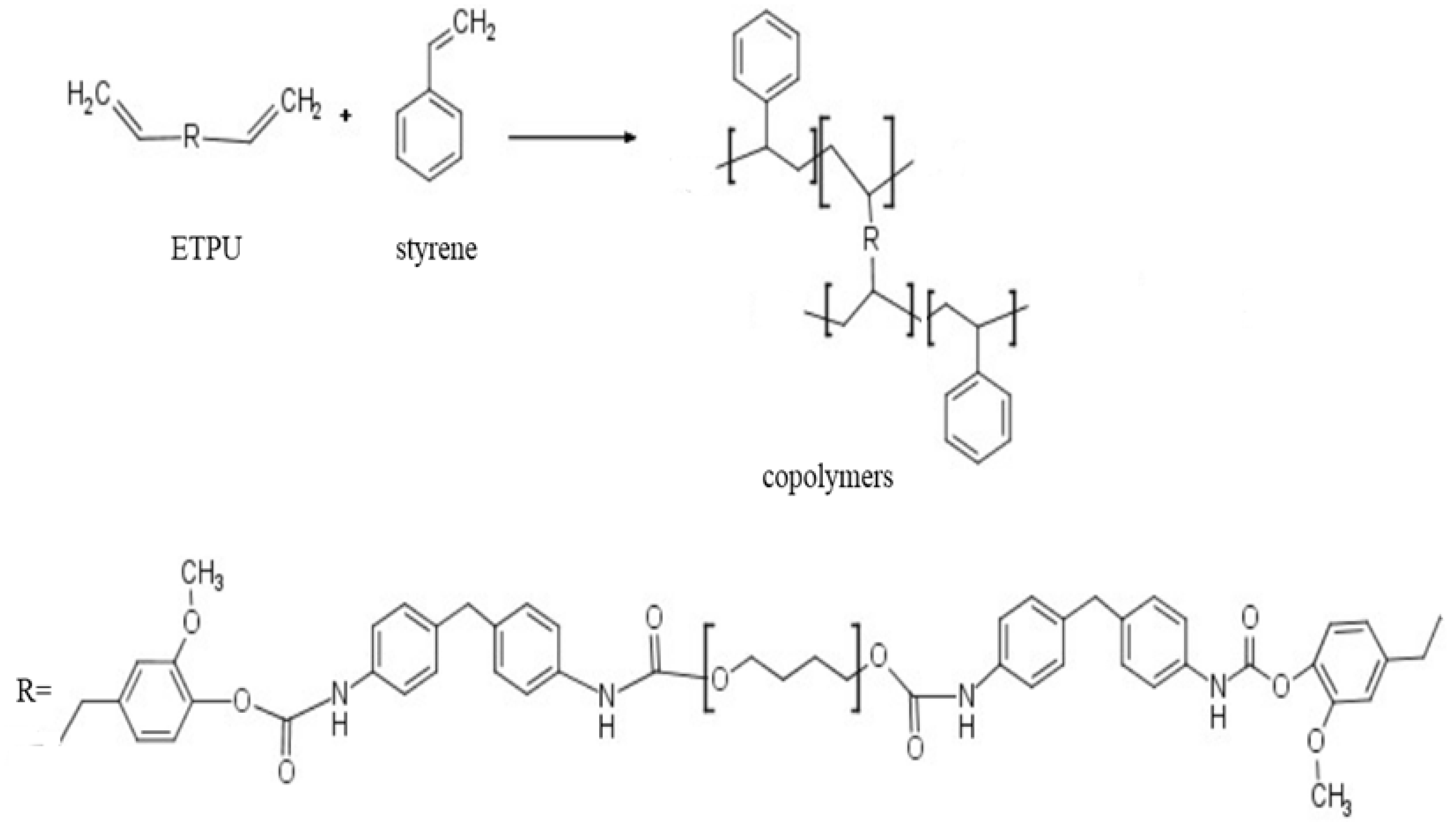
2.2. Synthesis of Eugenol-terminated Polyurethane (ETPU)
2.3. Preparation of Copolymers
2.4. Characterizations
3. Results and Discussion
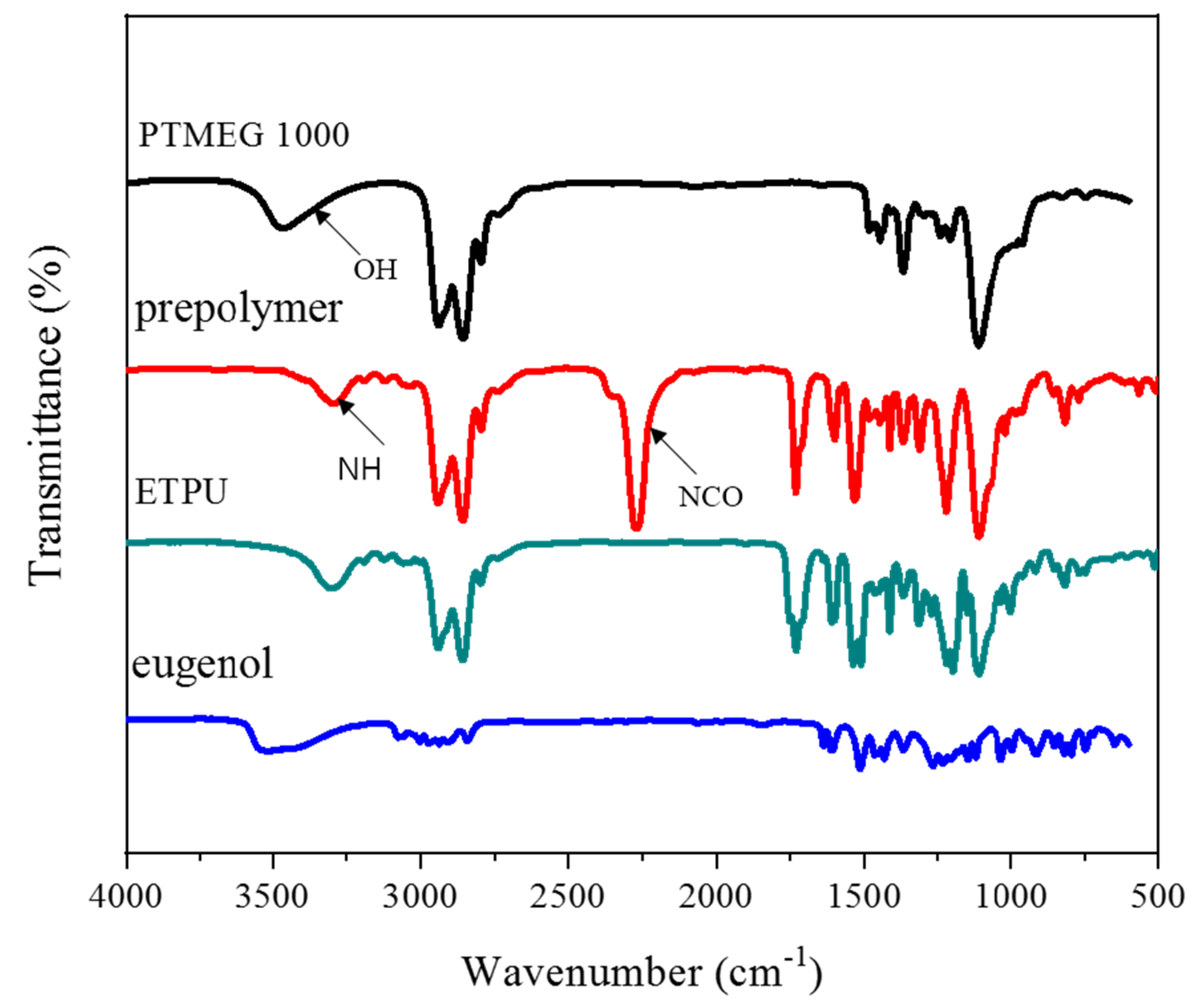
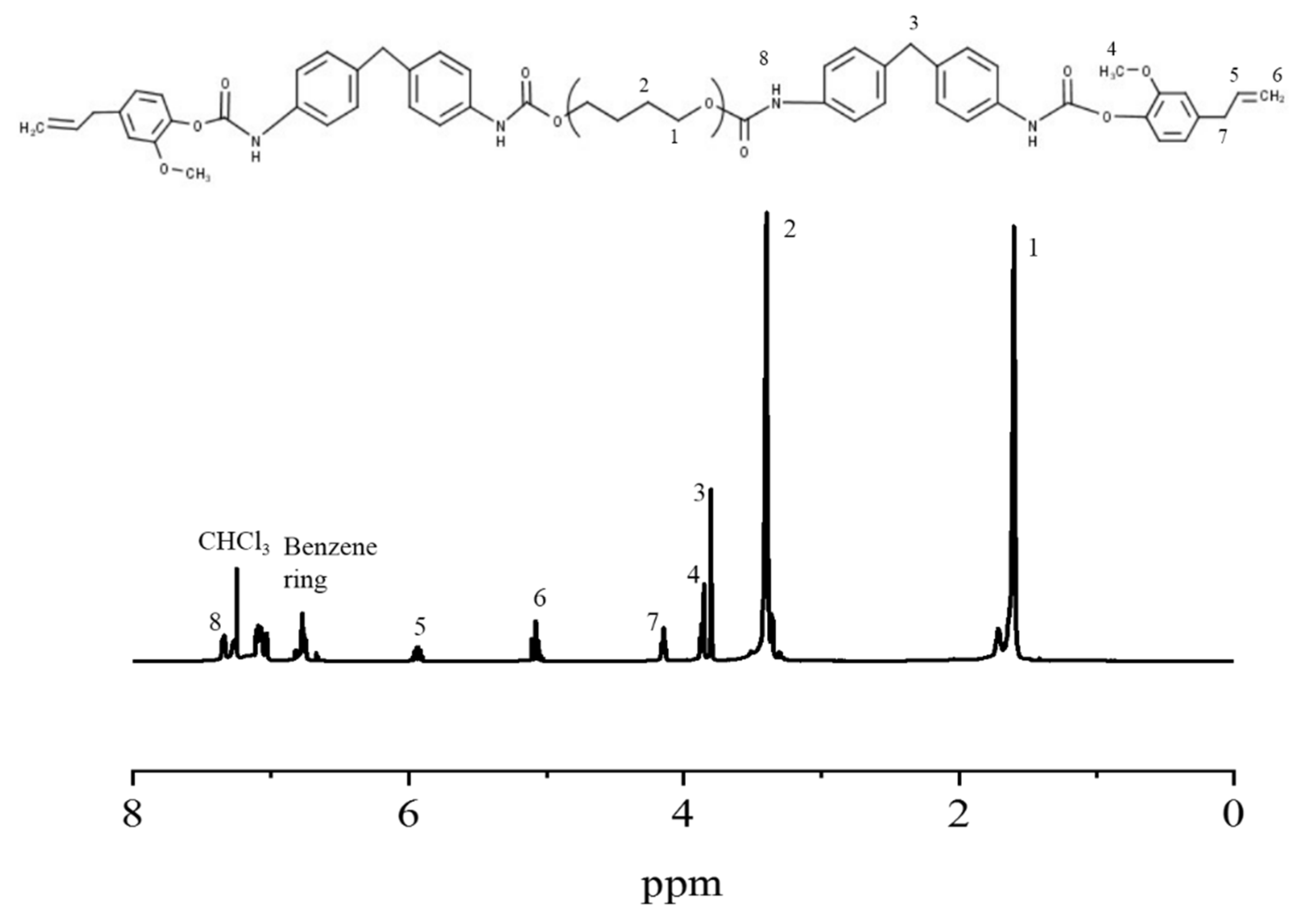
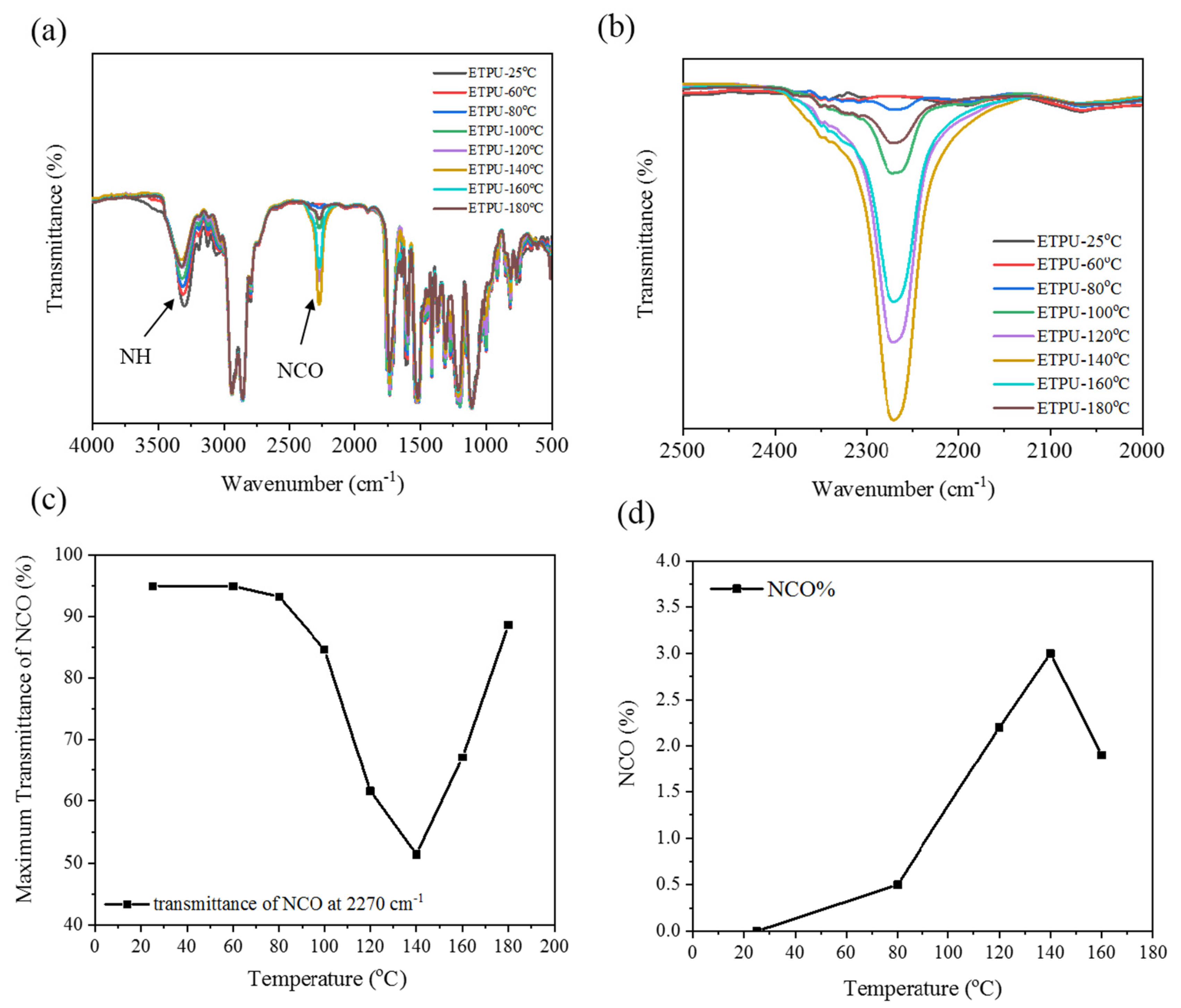
3.1. Synthesis and Characterization of ETPU
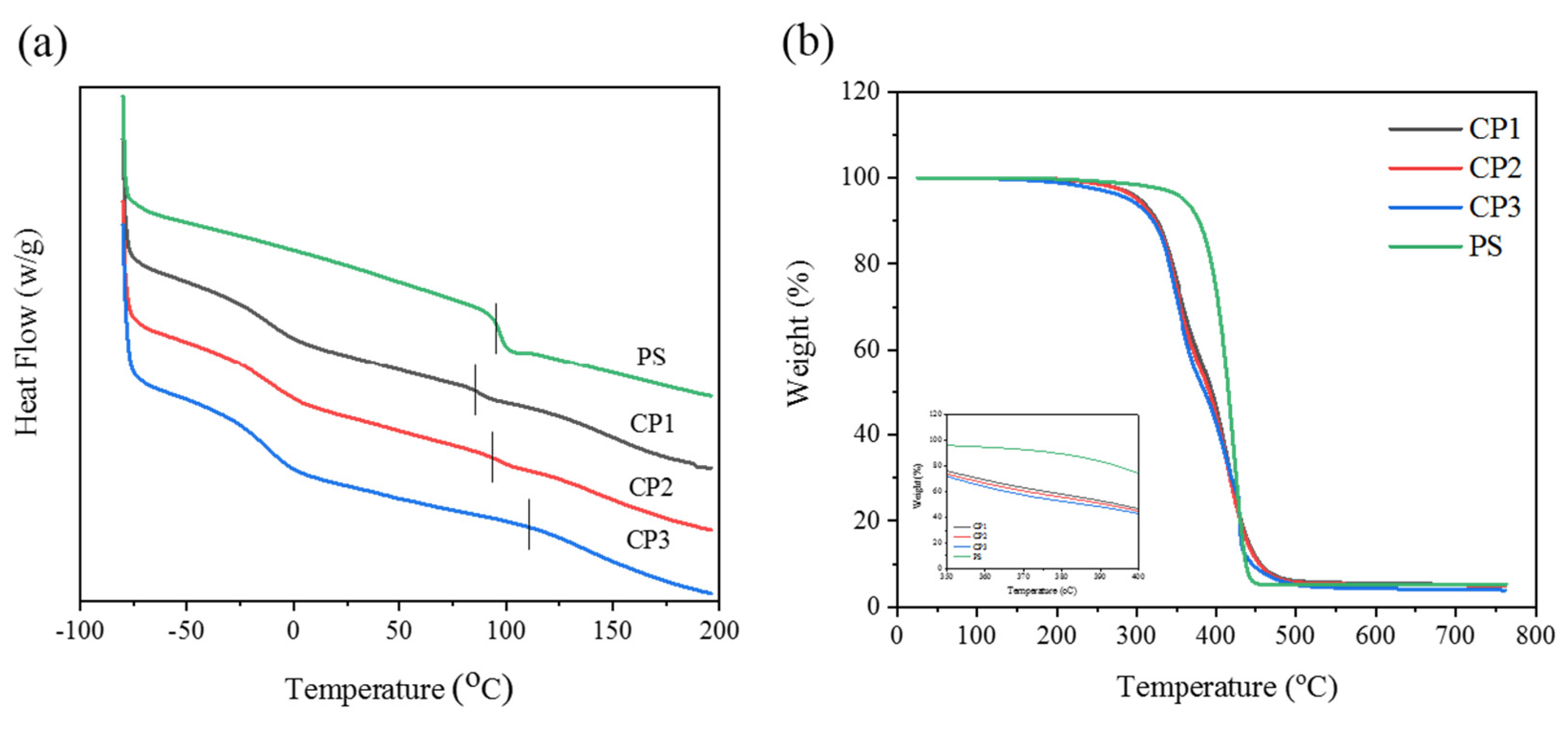
3.2. Thermal Stability of Copolymers
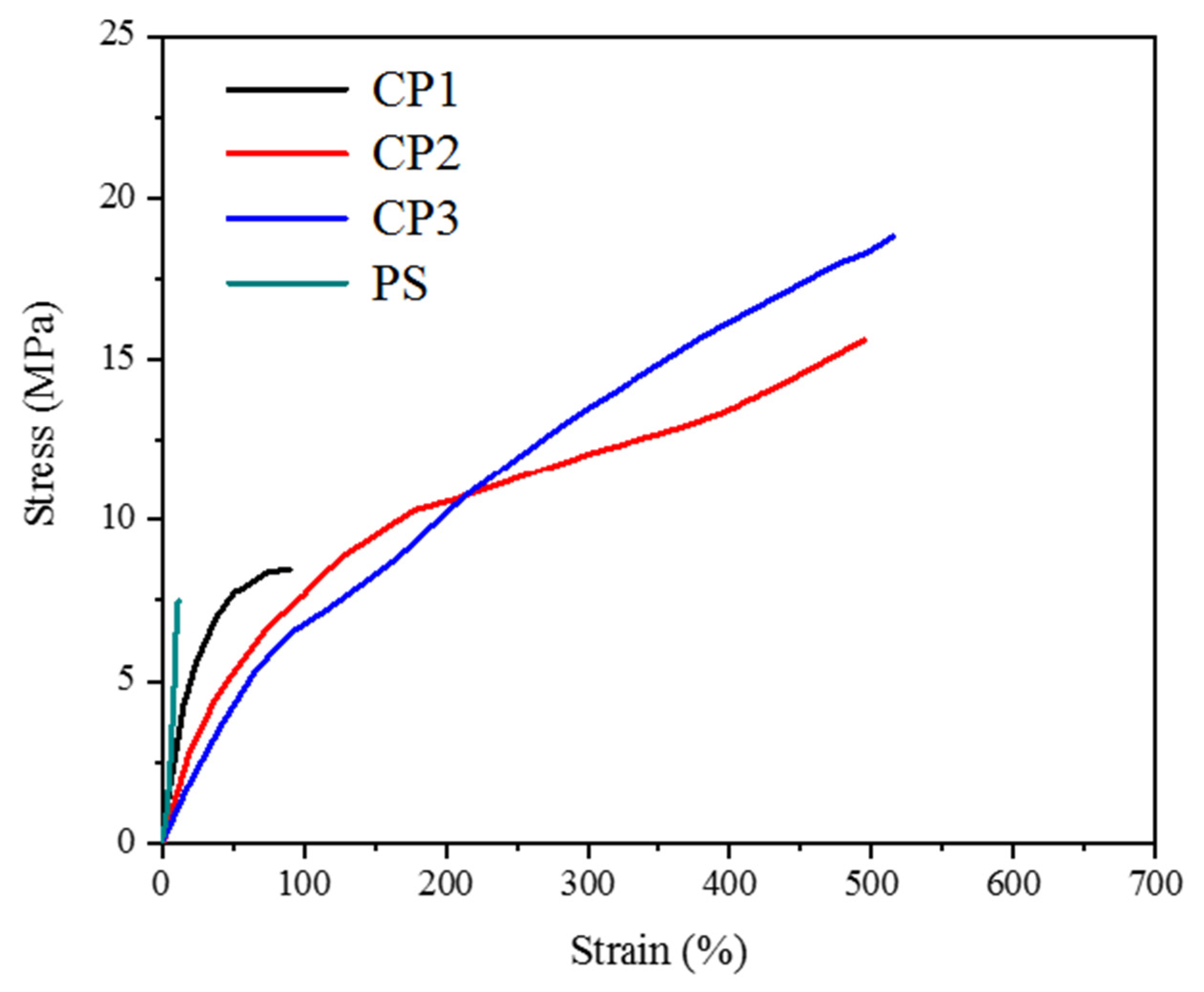
3.3. Mechanical Properties of the Copolymers
3.4. Dynamic Mechanical Properties
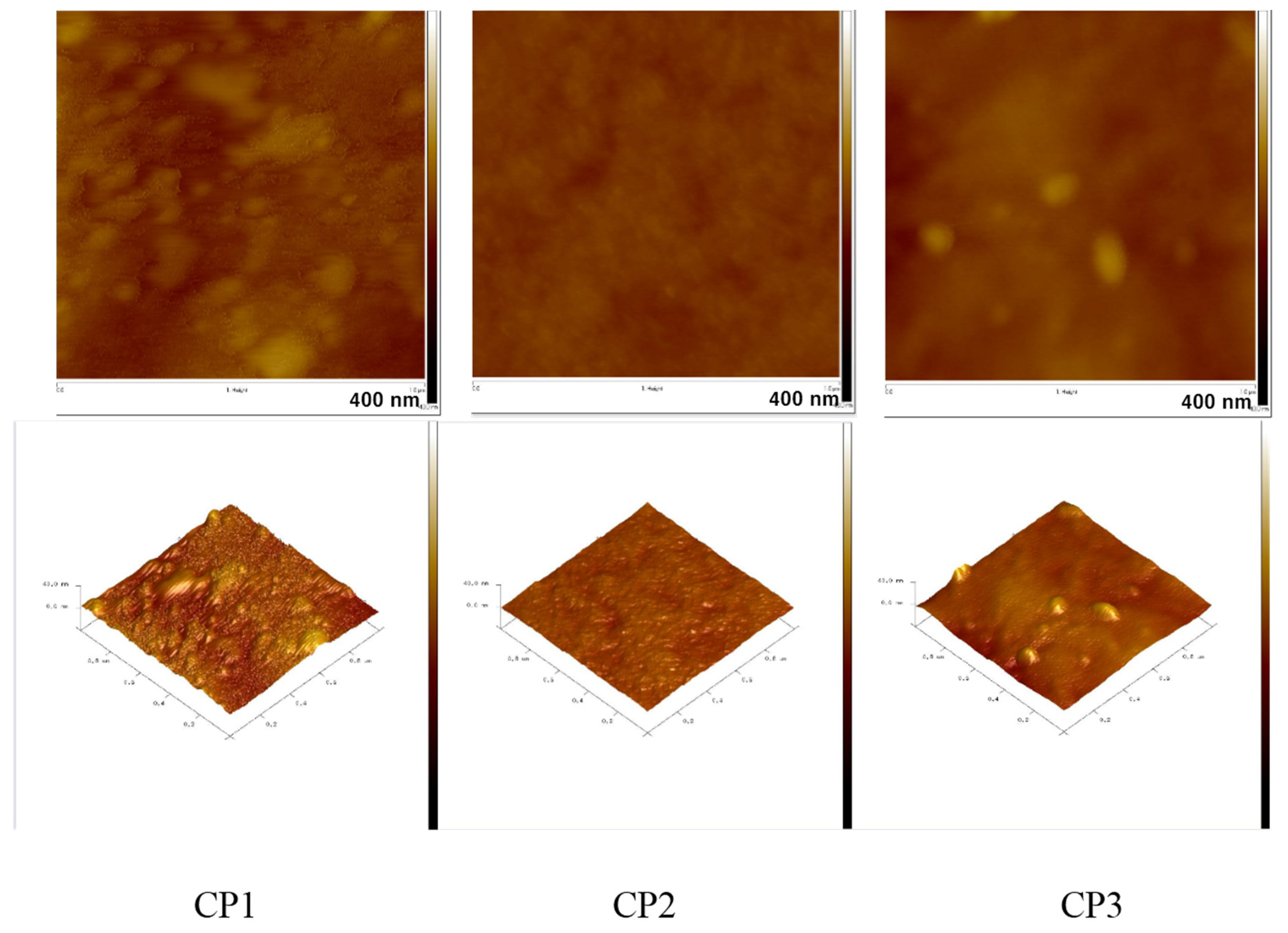
3.5. Atomic Force Microscopy of Copolymers
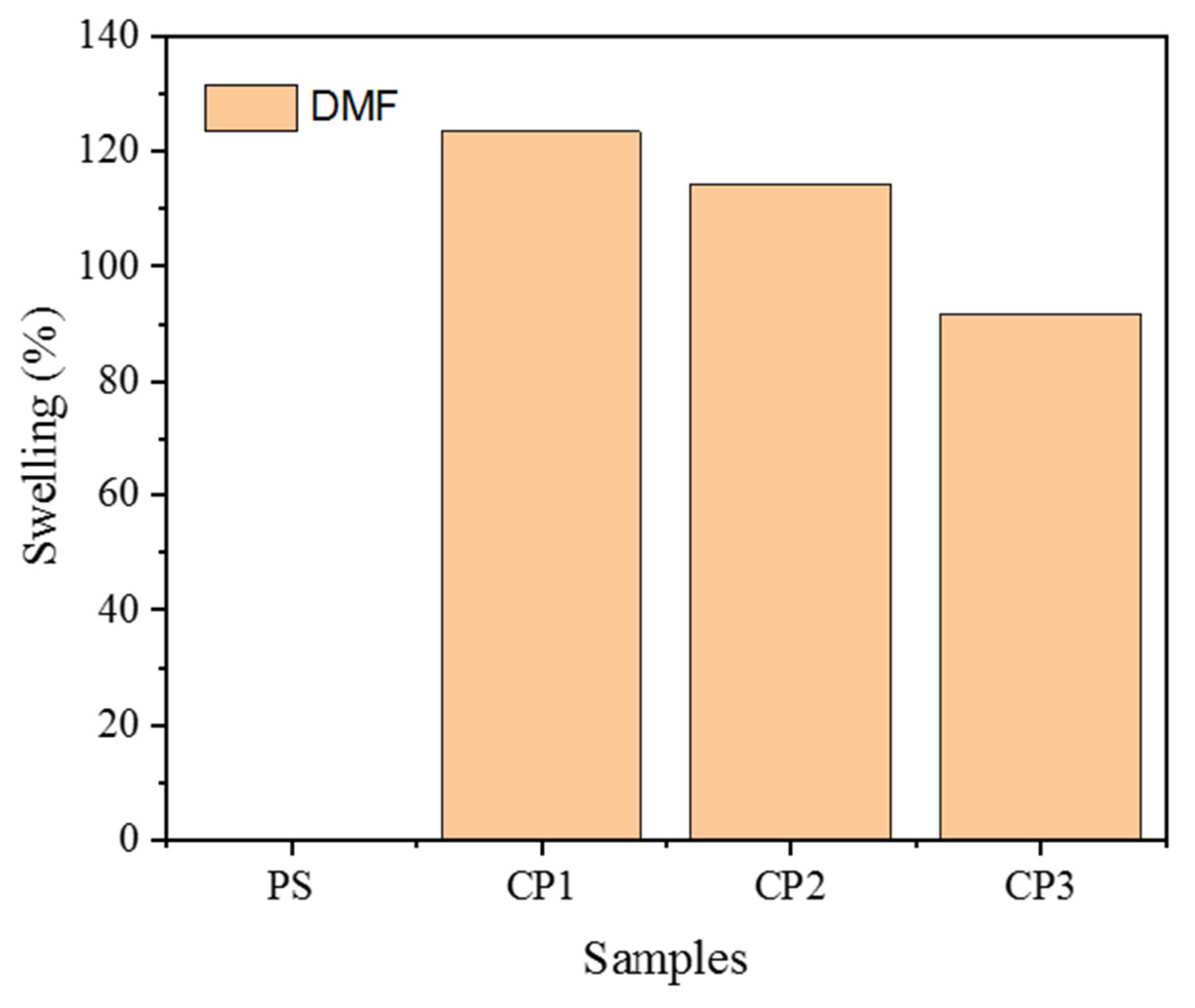
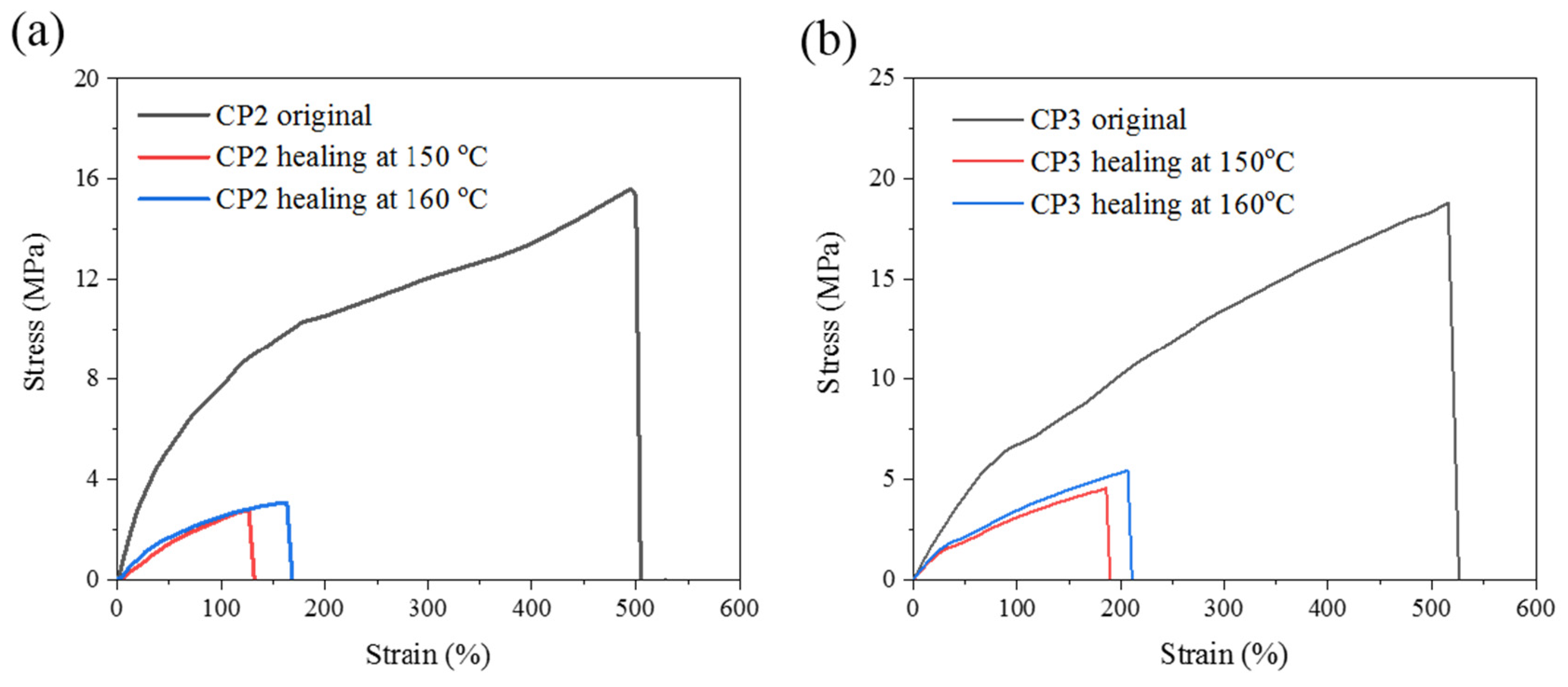
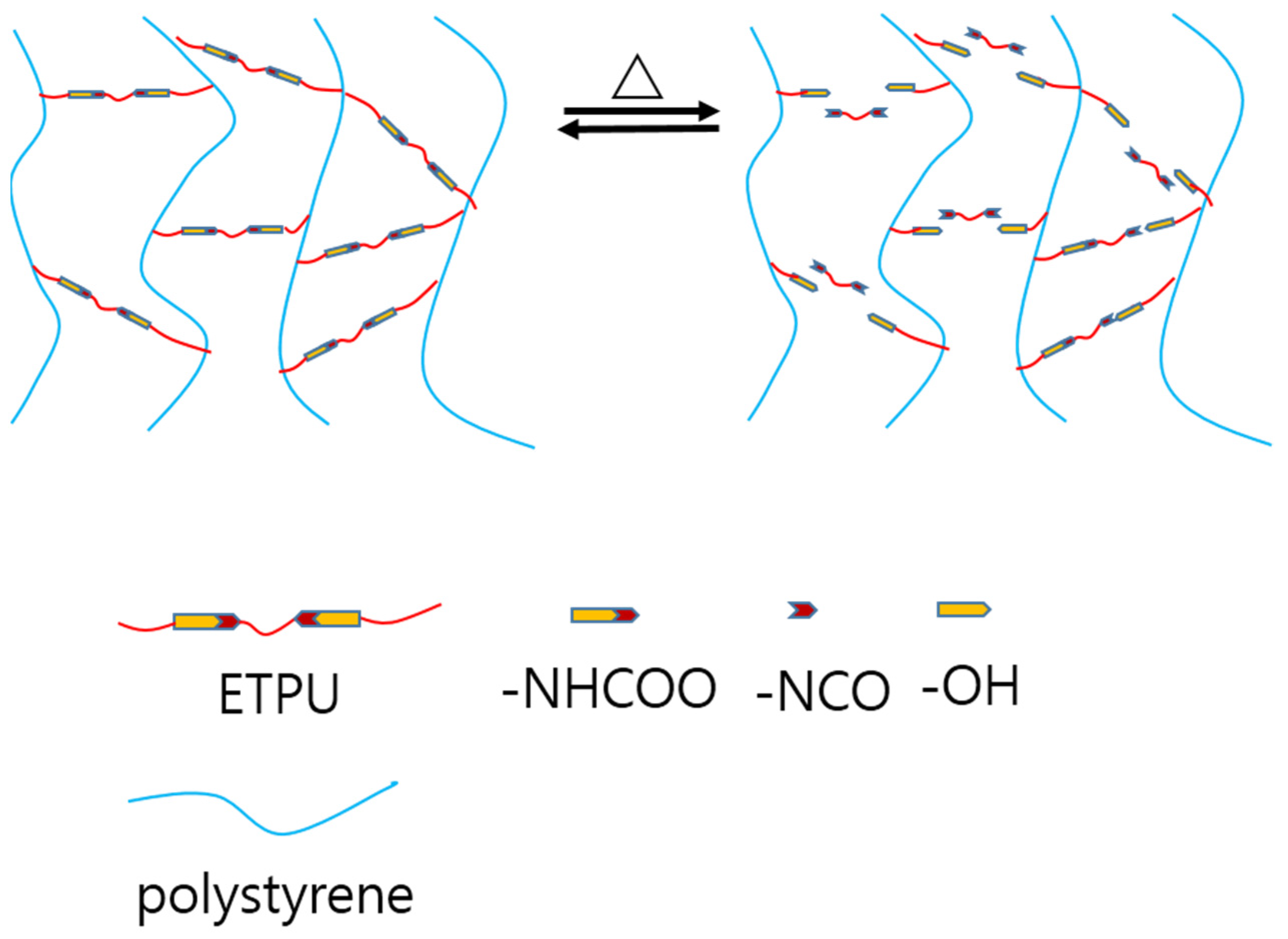
3.6. Self-healing Properties
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lligadas, G.; Ronda, J.C.; Galia, M.; Cadiz, V. Renewable polymeric materials from vegetable oils: Aprespective. Materials 2013, 16, 337–343. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, P.; Zhang, J.; Li, S.; Li, M.; Xia, J.; Zhou, Y. Preparation of biobased epoxies using tung oil fatty acid-derived C21 diacid and C22 triacid and the study of epoxy properties. Green Chem. 2013, 15, 2466–2475. [Google Scholar] [CrossRef]
- Llevot, A.; Grau, E.; Carlotti, S.; Grellier, S.; Cramail, H. From lignin-derived aromatic compounds to novel biobased polymers. Macromo. Papid Commum. 2016, 27, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Fache, M.; Viola, A.; Auvergne, R.; Boutevin, B.; Caillol, S. Biobased epoxy thermosets from vanillin-derived oligomers. Eur. Polym. J. 2015, 68, 526–535. [Google Scholar] [CrossRef]
- Inna, B.; Bretz, K.; Kabasci, S.; Kopitzky, R.; Springer, A. Succinic acid: A new platform chemical for biobased polymers from renewable resources. Chem. Eng. Technol. 2008, 31, 647–654. [Google Scholar]
- Thirukumaran, P.; Shakila, A.; Muthusamy, S. Synthesis and characterization of novel biobased benzoxazines form eugenol. Rsc Adv. 2014, 4, 7959–7966. [Google Scholar] [CrossRef]
- Zhang, M.Q.; Rong, M.Z. Design and synthesis of self-healing polymers. Sci. China Chem. 2012, 55, 648–676. [Google Scholar] [CrossRef]
- Chen, S.; Mahmood, N.; Beiner, M.; Binder, W.H. Self-healing materials from V-and H-shaped supramolecular architectures. Angew. Chem. Int. Ed. 2015, 54, 10188–10192. [Google Scholar] [CrossRef]
- Beck, J.B.; Multistimuli, S.J. Multiresponsive metallo-supramocular polymers. J. Am. Chem. Soc. 2003, 125, 13922–13923. [Google Scholar] [CrossRef]
- Cui, Y.L.; Tan, M.; Zhu, A.D.; Guo, M.Y. Non-covalent interaction cooperatively induced stretchy, tough and stimuli-responsive polyurethane–urea supramolecular (PUUS) hydrogels. J. Mater. Chem. B 2015, 3, 2834–2841. [Google Scholar] [CrossRef]
- Chang, K.; Jia, H.; Gu, S.Y. A transparent, highly strethchable, self-healing polyurethane based on disulfide bonds. Eur. Polym. J. 2019, 112, 822–831. [Google Scholar] [CrossRef]
- Yuan, C.E.; Rong, M.Z.; Zhang, M.Q.; Zhang, Z.P.; Yuan, Y.C. Self-healing of polymers via synchronous covalent bond fisson/radical recombination. Chem. Mater. 2011, 23, 5076–5081. [Google Scholar] [CrossRef]
- Deng, G.H.; Tang, C.M.; Li, H.Y.; Jiang, H.F.; Chen, Y.M. Covalent cross-linked polymer gels with reversible sol-gel transition and self-healing properties. Macromolecules 2010, 43, 1191–1194. [Google Scholar] [CrossRef]
- Oh, C.R.; Lee, D.I.; Park, J.H.; Lee, D.S. Thermally healale and recyclable graphene-nanoplate/epoxy composites via an in-situ Diels-Alder reaction on the graphene-nanoplate surface. Polymers 2019, 11, 1057. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.K.; Chul, O.; Chang, J.Y. The use of a thermally reversible bond for molecular imprinting of silica spheres. J. Am. Chem. Soc. 2002, 124, 14838–14839. [Google Scholar]
- Liu, T.; Hao, C.; Zhang, S.; Yang, X.N. A self-healable high glass transition temperature bioepoxy material based in vitrimer chemistry. Macromolecules 2018, 51, 5577–5585. [Google Scholar] [CrossRef]
- Ling, L.; Li, J.; Zhang, G.; Sun, R.; Wong, C.P. Self-healing and shape memory linear polyurethane based on disulfide linkages with excellent mechanical property. Macromol. Res. 2018, 26, 365–373. [Google Scholar] [CrossRef]
- Karami, Z.; Zohuriaan-Mehr, M.J.; Rostami, A. A bio-based thrmo-healable non-isocyanate polyurethane DA network in comparison with its epoxy counterpart. J. CO2 Util. 2017, 18, 294–302. [Google Scholar] [CrossRef]
- Zhu, K.; Song, Q.; Chen, H.; Hu, P. Thermally assisted self-healing polyurethane containing carboxyl groups. J. Appl. Polym. Sci. 2018, 135, 1–7. [Google Scholar] [CrossRef]
- Feng, L.; Yu, Z.; Bian, Y.; Lu, J.; Shi, X.; Chai, C. Self-healing behavior of polyurethanes based on dual actions of thermos-reversible Diels-Alder reaction and thermal movement of molecular chains. Polymer 2017, 124, 48–59. [Google Scholar] [CrossRef]
- Lin, C.; Sheng, D.; Liu, X.; Xu, S.; Ji, F.; Dong, L.; Zhou, Y.; Yang, Y. NIR induced self-healing electrical conductivity polyurethane/ graphene nanocomposites based on Diels-Alder reaction. Polymer 2018, 140, 150–157. [Google Scholar] [CrossRef]
- Kim, S.M.; Jeon, H.; Shin, S.H.; Park, S.A.; Jegal, J.; Heang, S.Y.; Oh, D.X.; Park, J. Superior toughness and fast self-healing at room temperature engineered by transparent elastomers. Adv. Mater. 2018, 30, 1705145. [Google Scholar] [CrossRef] [PubMed]
- Yangisawa, Y.; Nan, Y.; Okuro, K.; Aida, T. Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking. Science 2018, 359, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Kim, H.N.; Lee, D.S. Introduction of reversible urethane bonds based on vanilly alcohol for efficient self-healing of polyurethane elastomers. Molecules 2019, 24, 2201. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Li, S.H.; Li, M.; Xu, L.N.; Ding, H.Y.; Xia, J.L.; Huang, K. A thermal self-healing polyurethane thermoset based on phenolic urethane. Polym. J. 2017, 49, 775–781. [Google Scholar] [CrossRef]
- Simon, J.; Barla, F.; Kelemen-Haller, A.; Farkas, F.; Kraxner, M. Thermal stability of polyurethanes. Chromatographia 1988, 25, 99–106. [Google Scholar] [CrossRef]
- Telysheva, G.; Dobele, G.; Dizhbite, D.; Rossinska, G.; Jurkjane, V. Characterization of the transformations of lignocellulosic structures upon degradation in planted soil. J. Anal. Pyrol. 2007, 79, 52–60. [Google Scholar] [CrossRef]
- Philp, J. OECD policies for bioplastics in the content of a bioeconomy. Ind. Biotechnol. 2014, 10, 19–21. [Google Scholar] [CrossRef]
- Sun, L.; Singh, S.; Joo, M.; Simmones, B.A.; Auer, M. Non-invasive imaging of cellulose microfibril orientation within plant cell walls by polarized Raman microspectroscopy. Biotechnol. Bioeng. 2016, 113, 82–90. [Google Scholar] [CrossRef]
- Sabaa, M.W.; Mohamed, R.R. Organic thermal stabilizers for rigid poly (vinyl chloride). Part 8, Eugenol (4-allyl-2-methoxy-phenol). Polym. Degrad. Stab. 2007, 92, 587–595. [Google Scholar] [CrossRef]
- Harvey, B.G.; Sahagun, C.M.; Guenthner, A.J.; Groshens, T.J.; Cambrea, L.R.; Reams, J.T.; Mabry, J.M. A high-performance renewable thermosetting resin derived from eugenol. ChemSusChem 2014, 7, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Yang, S.; Teng, N.; Liu, Y.; Liu, X.; Zhu, J.; Zhao, J. Synthesis of eugenol-based silicon-containing benzoxazines and their applications as bio-based organic coatings. Coatings 2018, 8, 88. [Google Scholar] [CrossRef]
- Wan, J.T.; Gan, B.; Li, C.; Molina-Aldareguia, J.; Kalali, E.N.; Wang, X.; Wang, D.Y. A sustainable, eugenol-derived epoxy resin with high biobased content, modulus, hardness and low flammability: Synthesis, curing kinetics and structure-property relationship. Chem. Eng. 2016, 284, 1080–1093. [Google Scholar] [CrossRef]
- Maul, J.; Frushour, B.G.; Kontoff, J.R.; Eichenauer, H.; Ott, K.-H.; Schade, C. Polystyrene and Styrene Copolymers. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar] [CrossRef]
- Xiang, K.; Wang, X.; Huang, G.; Zheng, J.; Huang, J.; Li, G. Thermal ageing behavior of styrene-butadiene random copolymer: A study on the ageing mechanism and relaxation properties. Polym. Degrad Stab. 2012, 9, 1704–1715. [Google Scholar] [CrossRef]
- Xiang, K.; Wang, X.; Huang, G.; Zheng, J.; Huang, J.; Li, G. Thermogravimetric studies of styrene–butadiene rubber (SBR) after accelerated thermal aging. J. Anal. Calorim. 2014, 115, 247–254. [Google Scholar] [CrossRef]
- Munteanu, B.S.; Brebu, M.; Vasile, C. Thermal behaviour of binary and ternary copolymers containing acrylonitrile. Polym. Degrad Stab. 2013, 98, 1889–1897. [Google Scholar] [CrossRef]
- Adelia, F.F.M.; Mariana, A.S.; Vieira, M.G.A.; Marisa, M.B. Epoxidation of modified natural plasticizer obtained from rice fatty acids and application on polyvinylchloride films. J. Appl. Polym. Sci. 2013, 127, 3543–3549. [Google Scholar]
- Dave, V.J.; Patel, H.S. Synthesis and characterization of interpenetrating polymer networks from transesterified castor oil based polyurethane and polystyrene. J. Chem. Soc. 2017, 21, 18–24. [Google Scholar] [CrossRef]
- Quintana, R.; Persenaire, O.; Lemmouchi, Y.; Sampson, J.; Martin, S.; Bonnaud, L.; Dubois, P. Enhancement of cellulose acetate degradation under accelerated weathering by plasticization with ecofriendly plasticizers. Polym. Degrad Stab. 2013, 98, 1556–1562. [Google Scholar] [CrossRef]
- Klinger, M.; Tolbod, L.P.; Ogilby, P.R. Influence of a novel castroloil-derived additive on the mechanical properties and oxygen diffusivity of polystyrene. J. Appl. Polym. Sci. 2010, 118, 1643–1650. [Google Scholar]
- Gupta, A.P.; Ahmad, S.; Dev, A. Modification of novel bio-based resin-epoxidized soybean oil by conventional epoxy resin. Polym. Eng. Sci. 2011, 51, 1087–1091. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Codes | Monomer 1 Styrene (mol) | Monomer 2 ETPU (mol) | ETPU Content | |
|---|---|---|---|---|
| mol% | wt.% | |||
| PS | 10 | 0 | 0 | 0 |
| CP1 | 9.8 | 0.2 | 2 | 26.4 |
| CP2 | 9.6 | 0.4 | 4 | 42.3 |
| CP3 | 9.1 | 0.9 | 9 | 63.5 |
| Sample codes | Tensile Strength (MPa) | Elongation at Break (%) | Storage Modulus at 20 °C (MPa) | Tg (°C) | T d 5% (°C) | Residues at 700 °C (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| DMA | DSC | ||||||||
| Tgs | Tgh | Tgs | Tgh | ||||||
| PS | 7.1 | 10 | 2480 | 0 | 105 | 0 | 98 | 358 | 5.3 |
| CP1 | 8.4 | 80 | 138 | −16 | 101 | −11 | 90 | 303 | 5.2 |
| CP2 | 15.4 | 504 | 72 | −16 | 100 | −12 | 101 | 300 | 4.9 |
| CP3 | 18.8 | 600 | 71 | −16 | 114 | −12 | 109 | 290 | 3.9 |
| Sample | Original | After Healing | Healing Efficiency (%) | |||||
|---|---|---|---|---|---|---|---|---|
| 150 ℃ | 160 ℃ | 150 ℃ | 160 ℃ | |||||
| σ (MPa) | ε (%) | σ (MPa) | ε (%) | σ (MPa) | ε (%) | |||
| PS | 7.1 | 10 | - | - | - | - | - | - |
| CP1 | 8.4 | 80 | - | - | - | - | - | - |
| CP2 | 15.6 | 504 | 2.78 | 126 | 3.08 | 163 | 17.8 | 19.7 |
| CP3 | 18.8 | 600 | 4.6 | 190 | 5.5 | 206 | 24.5 | 29.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.-Y.; Shin, S.-R.; Lee, S.-H.; Lee, D.-S. Characteristics of Self-Healable Copolymers of Styrene and Eugenol Terminated Polyurethane Prepolymer. Polymers 2019, 11, 1674. https://doi.org/10.3390/polym11101674
Liang J-Y, Shin S-R, Lee S-H, Lee D-S. Characteristics of Self-Healable Copolymers of Styrene and Eugenol Terminated Polyurethane Prepolymer. Polymers. 2019; 11(10):1674. https://doi.org/10.3390/polym11101674
Chicago/Turabian StyleLiang, Jing-Yu, Se-Ra Shin, Soo-Hyoung Lee, and Dai-Soo Lee. 2019. "Characteristics of Self-Healable Copolymers of Styrene and Eugenol Terminated Polyurethane Prepolymer" Polymers 11, no. 10: 1674. https://doi.org/10.3390/polym11101674
APA StyleLiang, J. -Y., Shin, S. -R., Lee, S. -H., & Lee, D. -S. (2019). Characteristics of Self-Healable Copolymers of Styrene and Eugenol Terminated Polyurethane Prepolymer. Polymers, 11(10), 1674. https://doi.org/10.3390/polym11101674