Effects of Molecular Chain Length on the Contact Line Movement in Water/n-Alkane/Solid Systems



Abstract
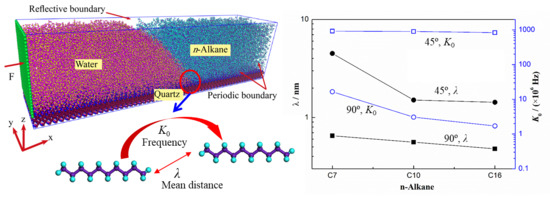
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Model and Methods
3. Results and Discussion
3.1. Density of Water and Alkane Molecules Near the Solid Surface
3.2. Tripping Force
3.3. Time-Averaged Velocity
3.4. Parameters of Molecular-Kinetics Theory
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Asserson, R.B.; Hoffmann, A.C.; Høiland, S.; Asvik, K.M. Interfacial tension measurement of freon hydrates by droplet deposition and contact angle measurements. J. Pet. Sci. Eng. 2009, 68, 209–217. [Google Scholar] [CrossRef]
- Mumley, T.E.; Radke, C.J.; Williams, M.C. Kinetics of liquid/liquid capillary rise: I. Experimental observations. J. Colloid Interface Sci. 1986, 109, 398–412. [Google Scholar] [CrossRef]
- Mumley, T.E.; Radke, C.J.; Williams, M.C. Kinetics of liquid/liquid capillary rise: II. Development and test of theory. J. Colloid Interface Sci. 1986, 109, 413–425. [Google Scholar] [CrossRef]
- Fermigier, M.; Jenffer, P. An experimental investigation of the dynamic contact angle in liquid-liquid systems. J. Colloid Interface Sci. 1991, 146, 226–241. [Google Scholar] [CrossRef]
- Foister, R.T. The kinetics of displacement wetting in liquid/liquid/solid systems. J. Colloid Interface Sci. 1990, 136, 266–282. [Google Scholar] [CrossRef]
- Voinov, O.V. Hydrodynamics of wetting. Fluid Dyn. 1976, 11, 714–721. [Google Scholar] [CrossRef]
- Petrov, J.G.; Radoev, B.P. Steady motion of the three phase contact line in model Langmuir-Blodgett systems. Colloid Polym. Sci. 1981, 259, 753–760. [Google Scholar] [CrossRef]
- Gennes, P.G.D. Wetting: Statics and dynamics. Rev. Mod. Phys. 1985, 57, 827–863. [Google Scholar] [CrossRef]
- Brochardwyart, F.; Gennes, P.G.D. Dynamics of partial wetting. Adv. Colloid Interface Sci. 1992, 39, 1–11. [Google Scholar] [CrossRef]
- Seveno, D.; Blake, T.D.; Goossens, S.; Coninck, J. De Predicting the wetting dynamics of a two-liquid system. Langmuir 2011, 27, 14958. [Google Scholar] [CrossRef]
- Goossens, S.; Seveno, D.; Rioboo, R.; Vaillant, A.; Conti, J.; Coninck, J.D. Can we predict the spreading of a two-liquid system from the spreading of the corresponding liquid-air systems? Langmuir 2011, 27, 9866–9872. [Google Scholar] [CrossRef] [PubMed]
- Ramiasa, M.; Ralston, J.; Fetzer, R.; Sedev, R. Contact Line Friction in Liquid-Liquid Displacement on Hydrophobic Surfaces. J. Phys. Chem. C 2011, 115, 24975–24986. [Google Scholar] [CrossRef]
- Fetzer, R.; Ramiasa, M.; Ralston, J. Dynamics of liquid-liquid displacement. Langmuir 2009, 25, 8069–8074. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.X.; Sun, C.Z.; Wen, B.Y.; Bai, B.F. Moving mechanisms of the three-phase contact line in a water-decane-silica system. RSC Adv. 2019, 9, 3092–3101. [Google Scholar] [CrossRef] [Green Version]
- Lichtfouse, É.; Eglinton, T.I. 13C and 14C evidence of pollution of a soil by fossil fuel and reconstruction of the composition of the pollutant. Org. Geochem. 1995, 23, 969–973. [Google Scholar] [CrossRef] [Green Version]
- Lichtfouse, É.; Bardoux, G.; Mariotti, A.; Balesdent, J.; Ballentine, D.C.; Macko, S.A. Molecular, 13C, and 14C evidence for the allochthonous and ancient origin of C16-C18 n -alkanes in modern soils. Geochim. Cosmochim. Acta 1997, 61, 1891–1898. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.G.; Xiang, Y.; Leng, Y.S. Computational simulations of solvation force and squeezing out of dodecane chain molecules in an atomic force microscope. J. Chem. Phys. 2017, 147, 054705. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, H.; Hess, D.W.; Breedveld, V. Effect of chain length on the wetting properties of alkyltrichlorosilane coated cellulose-based paper. Cellulose 2016, 23, 1401–1413. [Google Scholar] [CrossRef]
- Bertrand, E.; Blake, T.D.; De Coninck, J. Spreading dynamics of chain-like monolayers: A molecular dynamics study. Langmuir 2005, 21, 6628–6635. [Google Scholar] [CrossRef]
- Voue, M.; Rioboo, R.; Adao, M.H.; Conti, J.; Bondar, A.I.; Ivanov, D.A.; Blake, T.D.; De Coninck, J. Contact-line friction of liquid drops on self-assembled monolayers: Chain-length effects. Langmuir 2007, 23, 4695–4699. [Google Scholar] [CrossRef]
- Wen, B.Y.; Sun, C.Z.; Bai, B.F. Nanoparticle-induced ion-sensitive reduction in decane-water interfacial tension. Phys. Chem. Chem. Phys. 2018, 20, 22796–22804. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.Y.; Sun, C.Z.; Bai, B.F.; Gatapova, E.Y.; Kabov, O.A. Ionic hydration-induced evolution of decane-water interfacial tension. Phys. Chem. Chem. Phys. 2017, 19, 14606–14614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.Z.; Liu, M.; Bai, B.F. Molecular simulations on graphene-based membranes. Carbon 2019, 153, 481–494. [Google Scholar] [CrossRef]
- Sun, C.; Zhu, S.; Liu, M.; Shen, S.; Bai, B. Selective Molecular Sieving through a Large Graphene Nanopore with Surface Charges. J. Phys. Chem. Lett. 2019, 10, 7188–7194. [Google Scholar] [CrossRef] [PubMed]
- Bródka, A.; Zerda, T.W. Properties of liquid acetone in silica pores: Molecular dynamics simulation. J. Chem. Phys. 1996, 104, 6319–6326. [Google Scholar] [CrossRef]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.Z.; Bai, B.F. Gas diffusion on graphene surfaces. Phys. Chem. Chem. Phys. 2017, 19, 3894–3902. [Google Scholar] [CrossRef]
- Zheng, W.X.; Sun, C.Z.; Bai, B.F. Molecular Dynamics Study on the Effect of Surface Hydroxyl Groups on Three-Phase Wettability in Oil-Water-Graphite Systems. Polymers 2017, 9, 370. [Google Scholar] [CrossRef] [Green Version]
- Nagayama, G.; Cheng, P. Effects of interface wettability on microscale flow by molecular dynamics simulation. Int. J. Heat Mass Transf. 2004, 47, 501–513. [Google Scholar] [CrossRef]
- Emami, F.S.; Puddu, V.; Berry, R.J.; Varshney, V.; Patwardhan, S.V.; Perry, C.C.; Heinz, H. Force Field and a Surface Model Database for Silica to Simulate Interfacial Properties in Atomic Resolution. Chem. Mater. 2014, 26, 2647–2658. [Google Scholar] [CrossRef]
- Argyris, D.; Tummala, N.R.; Striolo, A.; Cole, D.R. Molecular structure and dynamics in thin water films at the silica and graphite surfaces. J. Phys. Chem. C 2008, 112, 13587–13599. [Google Scholar] [CrossRef]
- Wang, S.; Javadpour, F.; Feng, Q. Molecular dynamics simulations of oil transport through inorganic nanopores in shale. Fuel 2016, 171, 74–86. [Google Scholar] [CrossRef]
- Blake, T.D.; Haynes, J.M. Kinetics of Liquid/Liquid Displacement. J. Colloid Interface Sci. 1969, 30, 421–423. [Google Scholar] [CrossRef]
- Fetzer, R.; Ralston, J. Dynamic Dewetting Regimes Explored. J. Phys. Chem. C 2009, 113, 8888–8894. [Google Scholar] [CrossRef]
- Li, H.; Sedev, R.; Ralston, J. Dynamic wetting of a fluoropolymer surface by ionic liquids. Phys. Chem. Chem. Phys. 2011, 13, 3952–3959. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, W.; Sun, C.; Wen, B.; Bai, B.; Lichtfouse, E. Effects of Molecular Chain Length on the Contact Line Movement in Water/n-Alkane/Solid Systems. Polymers 2019, 11, 2081. https://doi.org/10.3390/polym11122081
Zheng W, Sun C, Wen B, Bai B, Lichtfouse E. Effects of Molecular Chain Length on the Contact Line Movement in Water/n-Alkane/Solid Systems. Polymers. 2019; 11(12):2081. https://doi.org/10.3390/polym11122081
Chicago/Turabian StyleZheng, Wenxiu, Chengzhen Sun, Boyao Wen, Bofeng Bai, and Eric Lichtfouse. 2019. "Effects of Molecular Chain Length on the Contact Line Movement in Water/n-Alkane/Solid Systems" Polymers 11, no. 12: 2081. https://doi.org/10.3390/polym11122081
APA StyleZheng, W., Sun, C., Wen, B., Bai, B., & Lichtfouse, E. (2019). Effects of Molecular Chain Length on the Contact Line Movement in Water/n-Alkane/Solid Systems. Polymers, 11(12), 2081. https://doi.org/10.3390/polym11122081