Facile Isolation of LCC-Fraction from Organosolv Lignin by Simple Soxhlet Extraction

 , ,
, ,  and
and 
Abstract
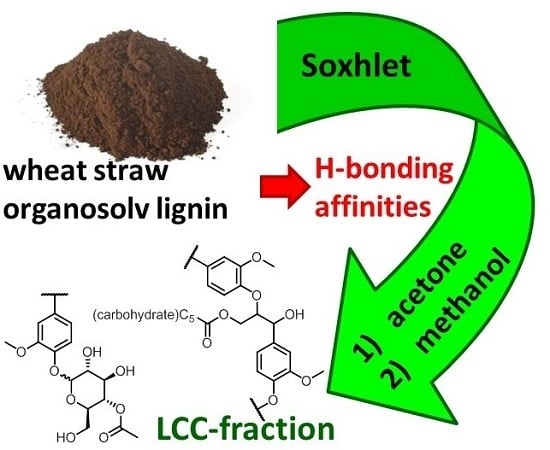
:
1. Introduction
2. Materials and Methods
2.1. General
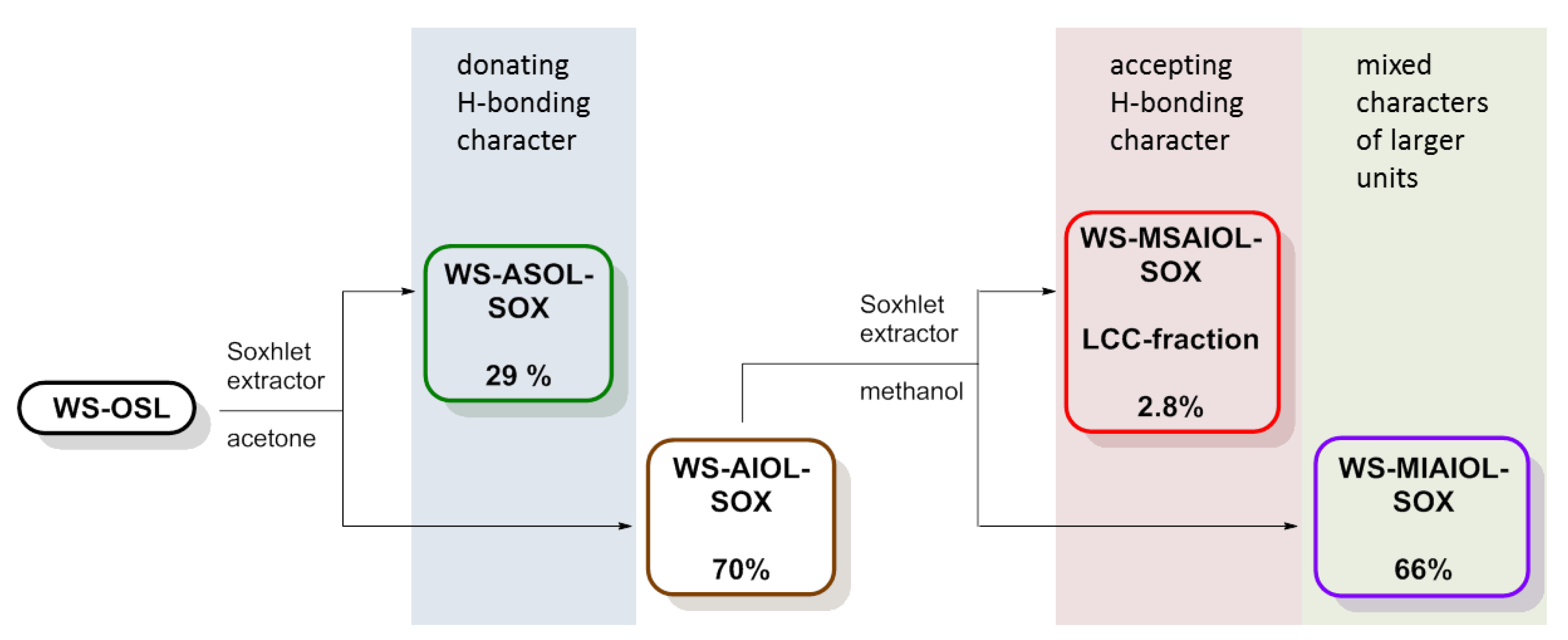
2.2. Soxhlet Fractionation of WS-OSL
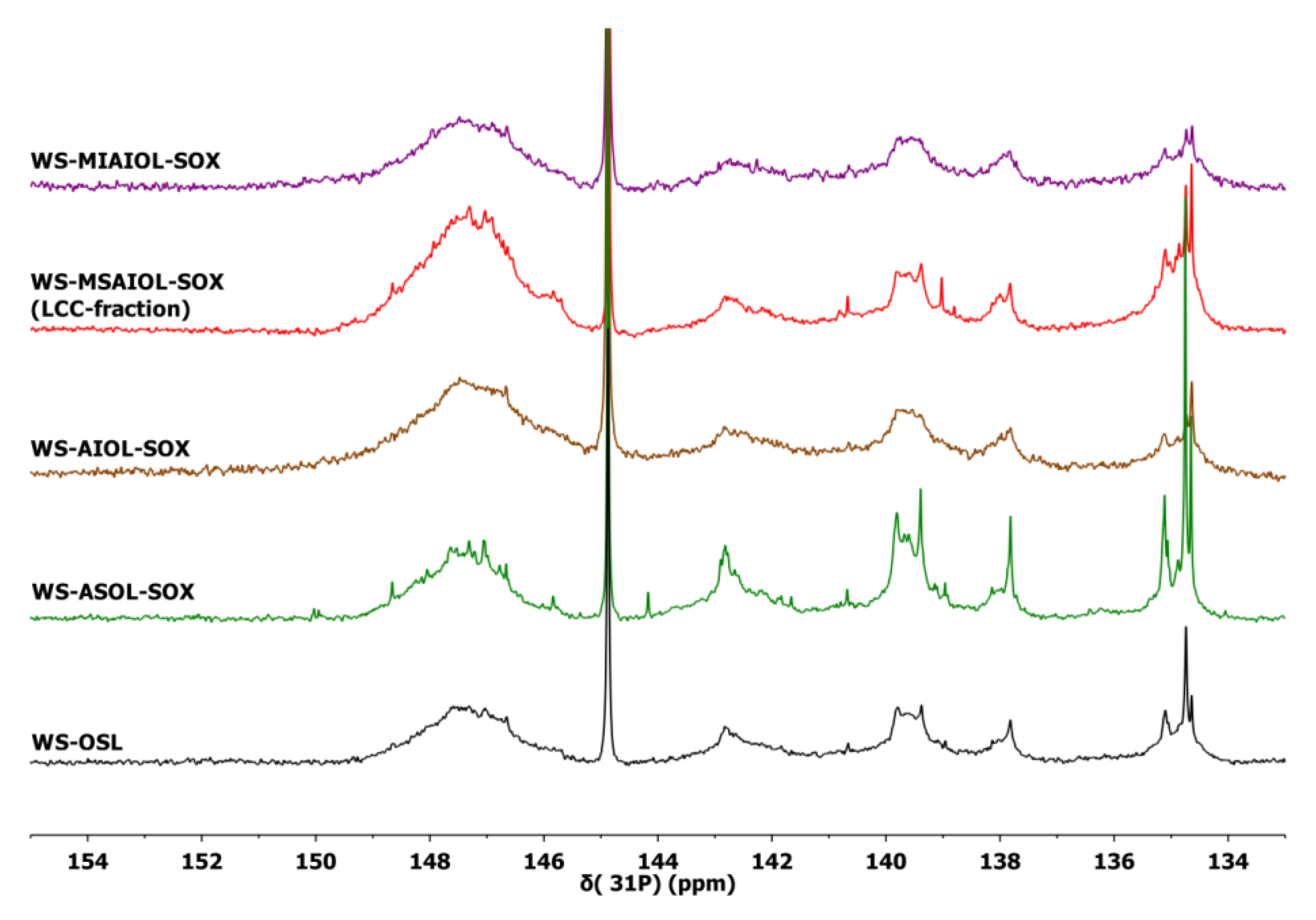
2.3. 31P NMR Analysis
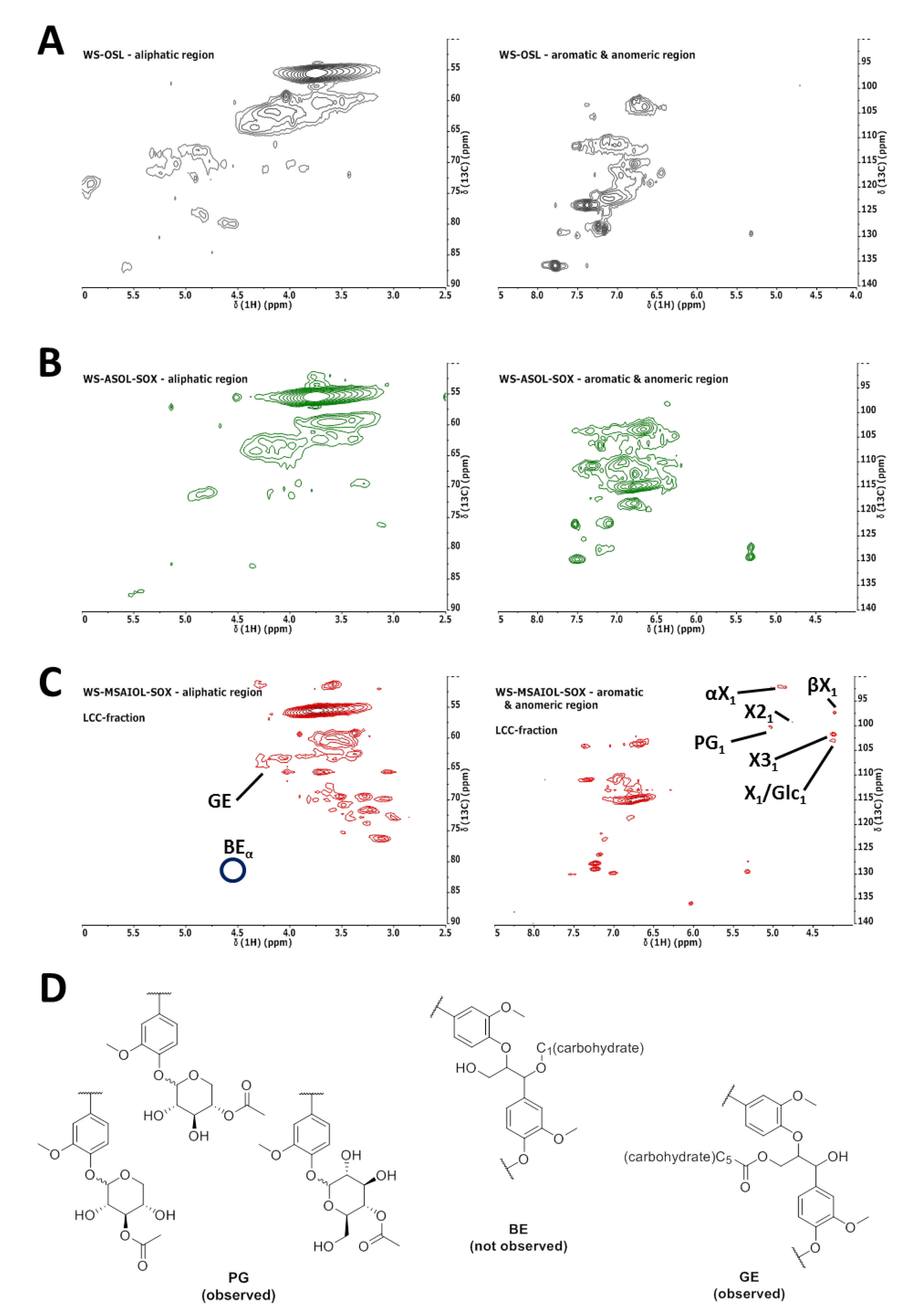
2.4. 1H−13C HSQC Measurements
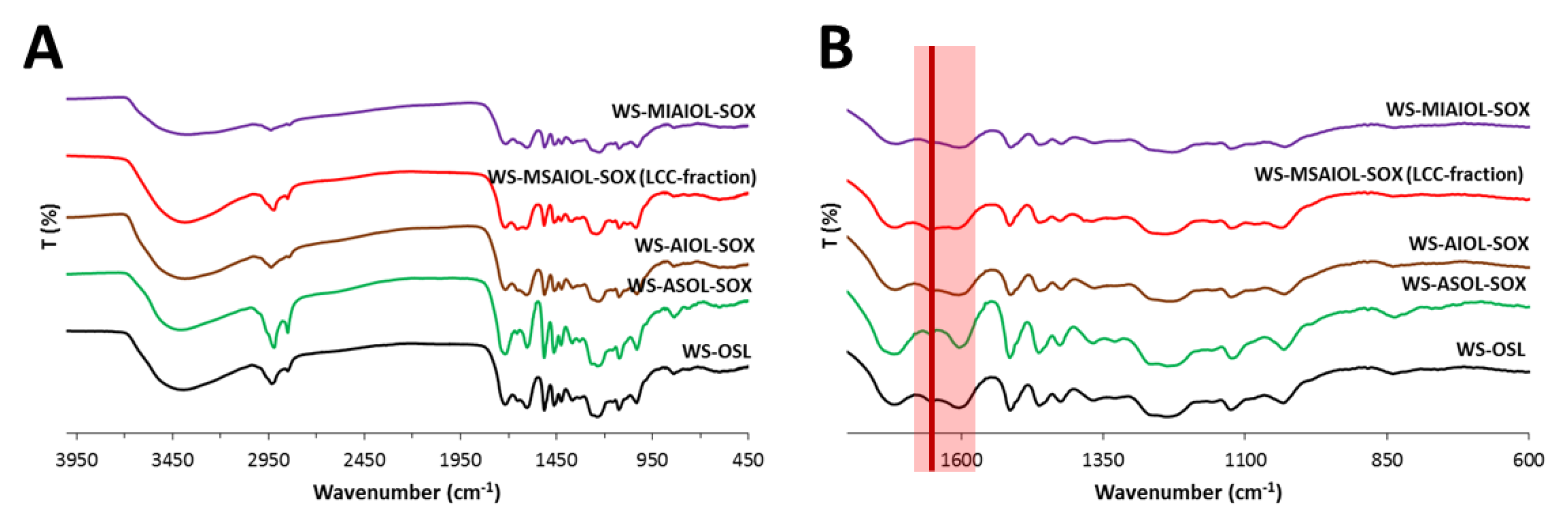
2.5. FT-IR Analysis
2.6. Gel Permeation Chromatographic Analyses
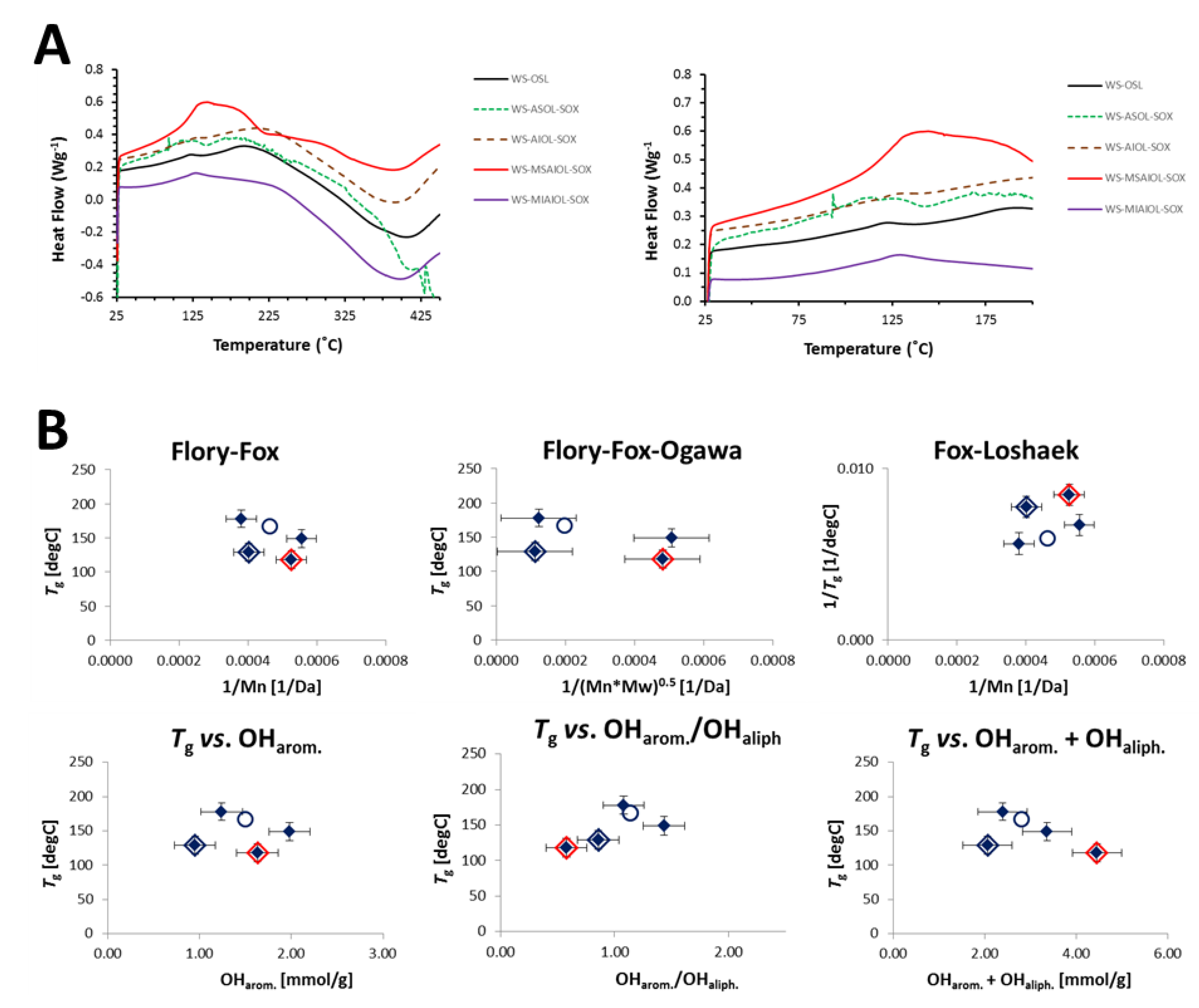
2.7. Differential Scanning Calorimetry
3. Results and Discussion
3.1. WS-OSL Fractionation
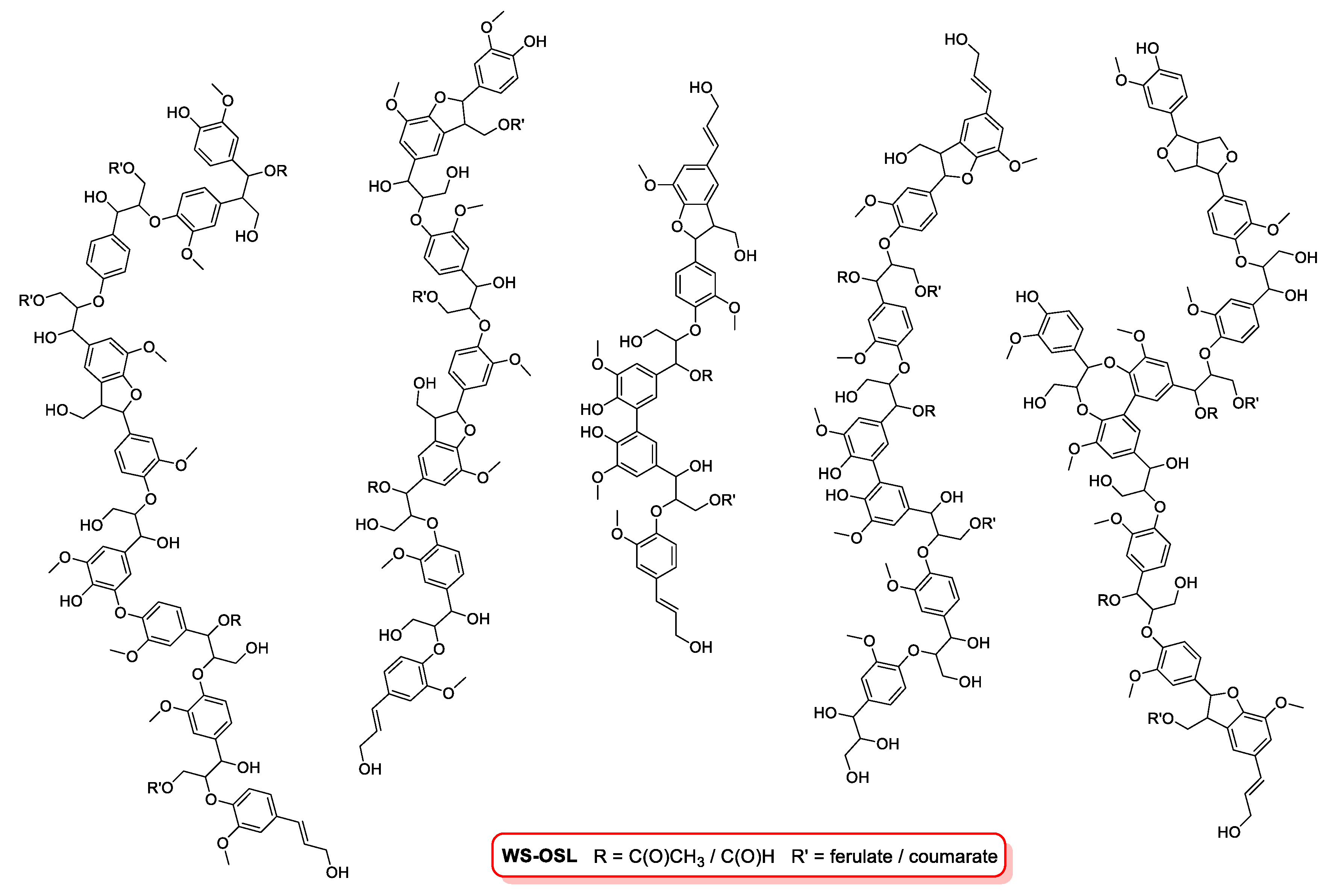
3.2. Structural Features of WS-OSL Fractions
3.3. Thermal Properties of Soxhlet-Derived WS-OSL Fractions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hu, T.Q. Chemcial Modification, Properties, and Usage of Lignin; Kluver Academic/plenum Publishers: New York, NY, USA, 2002. [Google Scholar]
- Glasser, W.G.; Northey, R.A.; Schultz, T.P. (Eds.) Lignin: Historical, Biological, and Materials Perspectives; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1999; Volume 742, ISBN 978-0-8412-3611-0. [Google Scholar]
- Glasser, W.G.; Sarkanen, S. Lignin: Properties and Materials; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1989. [Google Scholar]
- Argyropoulos, D.S. Materials, Chemicals, and Energy from Forest Biomass; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2007; ISBN 978-0-8412-3981-4. [Google Scholar]
- Lundquist, K.; Kirk, T.K. Fractionation-Purification of an Industrial Kraft Lignin. Tappi J. 1980, 63, 80–82. [Google Scholar]
- Schuerch, C. The Solvent Properties of Liquids and Their Relation to the Solubility, Swelling, Isolation and Fractionation of Lignin. J. Am. Chem. Soc. 1952, 74, 5061–5067. [Google Scholar] [CrossRef]
- Mousavioun, P.; Doherty, W.O.S. Chemical and thermal properties of fractionated bagasse soda lignin. Ind. Crops Prod. 2010, 31, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Sun, R.; Argyropoulos, D.S. Fractional Precipitation of Softwood Kraft Lignin: Isolation of Narrow Fractions Common to a Variety of Lignins. ACS Sustain. Chem. Eng. 2014, 2, 959–968. [Google Scholar] [CrossRef]
- Lange, H.; Schiffels, P.; Sette, M.; Sevastyanova, O.; Crestini, C. Fractional Precipitation of Wheat Straw Organosolv Lignin: Macroscopic Properties and Structural Insights. ACS Sustain. Chem. Eng. 2016, 4, 5136–5151. [Google Scholar] [CrossRef]
- Thring, R.W.; Vanderlaan, M.N.; Griffin, S.L. Fractionation of Alcell® Lignin by Sequential Solvent Extraction. J. Wood Chem. Technol. 1996, 16, 139–154. [Google Scholar] [CrossRef]
- Teng, N.-Y.; Dallmeyer, I.; Kadla, J.F. Effect of Softwood Kraft Lignin Fractionation on the Dispersion of Multiwalled Carbon Nanotubes. Ind. Eng. Chem. Res. 2013, 52, 6311–6317. [Google Scholar] [CrossRef]
- Gosselink, R.J.A.; van Dam, J.E.G.; de Jong, E.; Scott, E.L.; Sanders, J.P.M.; Li, J.; Gellerstedt, G. Fractionation, analysis, and PCA modeling of properties of four technical lignins for prediction of their application potential in binders. Holzforschung 2010, 64, 193–200. [Google Scholar] [CrossRef]
- Zikeli, F.; Ters, T.; Fackler, K.; Srebotnik, E.; Li, J. Successive and quantitative fractionation and extensive structural characterization of lignin from wheat straw. Ind. Crops Prod. 2014, 61, 249–257. [Google Scholar] [CrossRef]
- Duval, A.; Vilaplana, F.; Crestini, C.; Lawoko, M. Solvent screening for the fractionation of industrial kraft lignin. Holzforschung 2015, 70, 11–20. [Google Scholar] [CrossRef]
- Griffini, G.; Passoni, V.; Suriano, R.; Levi, M.; Turri, S. Polyurethane Coatings Based on Chemically Unmodified Fractionated Lignin. ACS Sustain. Chem. Eng. 2015, 3, 1145–1154. [Google Scholar] [CrossRef]
- Passoni, V.; Scarica, C.; Levi, M.; Turri, S.; Griffini, G. Fractionation of Industrial Softwood Kraft Lignin: Solvent Selection as a Tool for Tailored Material Properties. ACS Sustain. Chem. Eng. 2016, 4, 2232–2242. [Google Scholar] [CrossRef]
- Hernes, P.J.; Robinson, A.C.; Aufdenkampe, A.K. Fractionation of lignin during leaching and sorption and implications for organic matter “freshness”. Geophys. Res. Lett. 2007, 34, L17401. [Google Scholar] [CrossRef]
- Toledano, A.; García, A.; Mondragon, I.; Labidi, J. Lignin separation and fractionation by ultrafiltration. Sep. Purif. Technol. 2010, 71, 38–43. [Google Scholar] [CrossRef]
- Sevastyanova, O.; Helander, M.; Chowdhury, S.; Lange, H.; Wedin, H.; Zhang, L.; Ek, M.; Kadla, J.F.; Crestini, C.; Lindström, M.E. Tailoring the molecular and thermo–mechanical properties of kraft lignin by ultrafiltration. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Duval, A.; Molina-Boisseau, S.; Chirat, C. Fractionation of lignosulfonates: Comparison of ultrafiltration and ethanol solubility to obtain a set of fractions with distinct properties. Holzforschung 2014, 69, 127–134. [Google Scholar] [CrossRef]
- Rohde, V.; Böringer, S.; Tübke, B.; Adam, C.; Dahmen, N.; Schmiedl, D. Fractionation of three different lignins by thermal separation techniques—A comparative study. GCB Bioenergy 2018, 1–12. [Google Scholar] [CrossRef]
- Erdmann, J. Ueber die Concretionen in den Birnen. Ann. Chem. Pharm. 1866, 138, 1–19. [Google Scholar] [CrossRef]
- Tarasov, D.; Leitch, M.; Fatehi, P. Lignin–carbohydrate complexes: Properties, applications, analyses, and methods of extraction: A review. Biotechnol. Biofuels 2018, 11, 269. [Google Scholar] [CrossRef]
- Koshijima, T.; Watanabe, T. Association Between Lignin and Carbohydrates in Wood and Other Plant Tissues; Springer Series in Wood Science; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2003; ISBN 978-3-642-07853-8. [Google Scholar]
- Yuan, T.-Q.; Sun, S.-N.; Xu, F.; Sun, R.-C. Characterization of Lignin Structures and Lignin–Carbohydrate Complex (LCC) Linkages by Quantitative 13C and 2D HSQC NMR Spectroscopy. J. Agric. Food Chem. 2011, 59, 10604–10614. [Google Scholar] [CrossRef]
- Zikeli, F.; Ters, T.; Fackler, K.; Srebotnik, E.; Li, J. Wheat straw lignin fractionation and characterization as lignin-carbohydrate complexes. Ind. Crops Prod. 2016, 85, 309–317. [Google Scholar] [CrossRef]
- Delmas, M.; Benjelloun, M. Process for the Separation of Lignins and Sugars from an Extraction Liquor. EP2580246B1, 7 March 2018. [Google Scholar]
- Tachon, N.; Benjelloun-Mlayah, B.; Delmas, M. Organosolv Wheat Straw Lignin as a Phenol Substitute for Green Phenolic Resins. BioResources 2016, 11, 5797–5815. [Google Scholar] [CrossRef]
- Mbotchak, L.; Le Morvan, C.; Duong, K.L.; Rousseau, B.; Tessier, M.; Fradet, A. Purification, Structural Characterization, and Modification of Organosolv Wheat Straw Lignin. J. Agric. Food Chem. 2015, 63, 5178–5188. [Google Scholar] [CrossRef] [PubMed]
- Cachet, N.; Camy, S.; Benjelloun-Mlayah, B.; Condoret, J.-S.; Delmas, M. Esterification of organosolv lignin under supercritical conditions. Ind. Crops Prod. 2014, 58, 287–297. [Google Scholar] [CrossRef]
- Snelders, J.; Dornez, E.; Benjelloun-Mlayah, B.; Huijgen, W.J.J.; de Wild, P.J.; Gosselink, R.J.A.; Gerritsma, J.; Courtin, C.M. Biorefining of wheat straw using an acetic and formic acid based organosolv fractionation process. Bioresour. Technol. 2014, 156, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Delmas, G.-H.; Benjelloun-Mlayah, B.; Bigot, Y.L.; Delmas, M. Functionality of wheat straw lignin extracted in organic acid media. J. Appl. Polym. Sci. 2011, 121, 491–501. [Google Scholar] [CrossRef]
- Giannì, P.; Lange, H.; Crestini, C. Lipoxygenase: Unprecedented Carbon-Centered Lignin Activation. ACS Sustain. Chem. Eng. 2018, 6, 5085–5096. [Google Scholar] [CrossRef]
- Granata, A.; Argyropoulos, D.S. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a Reagent for the Accurate Determination of the Uncondensed and Condensed Phenolic Moieties in Lignins. J. Agric. Food Chem. 1995, 43, 1538–1544. [Google Scholar] [CrossRef]
- Jiang, Z.-H.; Argyropoulos, D.S.; Granata, A. Correlation analysis of 31P NMR chemical shifts with substituent effects of phenols. Magn. Reson. Chem. 1995, 33, 375–382. [Google Scholar] [CrossRef]
- Argyropoulos, D.S. 31P NMR in Wood Chemistry: A Review of Recent Progress. Res. Chem. Intermed. 1995, 21, 373–395. [Google Scholar] [CrossRef]
- Lange, H.; Rulli, F.; Crestini, C. Gel Permeation Chromatography in Determining Molecular Weights of Lignins: Critical Aspects Revisited for Improved Utility in the Development of Novel Materials. ACS Sustain. Chem. Eng. 2016, 4, 5167–5180. [Google Scholar] [CrossRef]
- Lu, F.; Ralph, J. DFRC Method for Lignin Analysis. 1. New Method for β-Aryl Ether Cleavage: Lignin Model Studies. J. Agric. Food Chem. 1997, 45, 4655–4660. [Google Scholar] [CrossRef]
- You, T.-T.; Zhang, L.-M.; Zhou, S.-K.; Xu, F. Structural elucidation of lignin–carbohydrate complex (LCC) preparations and lignin from Arundo donax Linn. Ind. Crops Prod. 2015, 71, 65–74. [Google Scholar] [CrossRef]
- Crestini, C.; Argyropoulos, D.S. Structural Analysis of Wheat Straw Lignin by Quantitative 31P and 2D NMR Spectroscopy. The Occurrence of Ester Bonds and α-O-4 Substructures. J. Agric. Food Chem. 1997, 45, 1212–1219. [Google Scholar] [CrossRef]
- Del Río, J.C.; Rencoret, J.; Prinsen, P.; Martínez, Á.T.; Ralph, J.; Gutiérrez, A. Structural Characterization of Wheat Straw Lignin as Revealed by Analytical Pyrolysis, 2D-NMR, and Reductive Cleavage Methods. J. Agric. Food Chem. 2012, 60, 5922–5935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, T.G., Jr.; Flory, P.J. Second-Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar]
- Ogawa, T. Effects of molecular weight on mechanical properties of polypropylene. J. Appl. Polym. Sci. 1992, 44, 1869–1871. [Google Scholar] [CrossRef] [Green Version]
- Fox, T.G.; Loshaek, S. Influence of molecular weight and degree of crosslinking on the specific volume and glass temperature of polymers. J. Polym. Sci. 1955, 15, 371–390. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Sample | Yield (%) | Mn (Da) | Mw (Da) | PDI a |
|---|---|---|---|---|---|
| 1 | WS-OSL | --- | 2200 | 12,100 | 5.6 |
| 2 | WS-ASOL-SOX | 29 | 1800 | 2200 | 1.2 |
| 3 | WS-AIOL-SOX | 70 | 2600 | 25,200 | 9.6 |
| 4 | WS-MSAIOL-SOX (LCC-fraction) | 2.8 | 1900 | 2300 | 1.2 |
| 5 | WS-MIAIOL-SOX | 66 | 2500 | 32,000 | >10 |
| Entry | Sample | Aliphatic OH (mmol/g) | Aromatic OH (mmol/g) | Acidic OH (mmol/g) | Total OH (mmol/g) | ||
|---|---|---|---|---|---|---|---|
| Siringyl | Guaiacyl | p-hydroxy | |||||
| 1 | WS-OSL | 1.31 | 0.63 | 0.59 | 0.27 | 1.48 | 4.28 |
| 2 | WS-ASOL-SOX | 1.38 | 0.85 | 0.82 | 0.31 | 1.99 | 5.35 |
| 3 | WS-AIOL-SOX | 1.15 | 0.64 | 0.39 | 0.21 | 1.24 | 3.63 |
| 4 | WS-MSAIOL-SOX (LCC-fraction) | 2.82 | 0.66 | 0.67 | 0.30 | 1.63 | 6.08 |
| 5 | WS-MIAIOL-SOX | 1.11 | 0.37 | 0.38 | 0.20 | 0.95 | 3.01 |
| Sample | OSL | ASOL-SOX | AIOL-SOX | MSAIOL-SOX (LCC-fraction) | MIAIOL-SOX |
|---|---|---|---|---|---|
| Tg (°C) a | 168 | 149 | 178 | 118 | 129 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebrahimi Majdar, R.; Ghasemian, A.; Resalati, H.; Saraeian, A.; Crestini, C.; Lange, H. Facile Isolation of LCC-Fraction from Organosolv Lignin by Simple Soxhlet Extraction. Polymers 2019, 11, 225. https://doi.org/10.3390/polym11020225
Ebrahimi Majdar R, Ghasemian A, Resalati H, Saraeian A, Crestini C, Lange H. Facile Isolation of LCC-Fraction from Organosolv Lignin by Simple Soxhlet Extraction. Polymers. 2019; 11(2):225. https://doi.org/10.3390/polym11020225
Chicago/Turabian StyleEbrahimi Majdar, Reza, Ali Ghasemian, Hossein Resalati, Ahmadreza Saraeian, Claudia Crestini, and Heiko Lange. 2019. "Facile Isolation of LCC-Fraction from Organosolv Lignin by Simple Soxhlet Extraction" Polymers 11, no. 2: 225. https://doi.org/10.3390/polym11020225
APA StyleEbrahimi Majdar, R., Ghasemian, A., Resalati, H., Saraeian, A., Crestini, C., & Lange, H. (2019). Facile Isolation of LCC-Fraction from Organosolv Lignin by Simple Soxhlet Extraction. Polymers, 11(2), 225. https://doi.org/10.3390/polym11020225